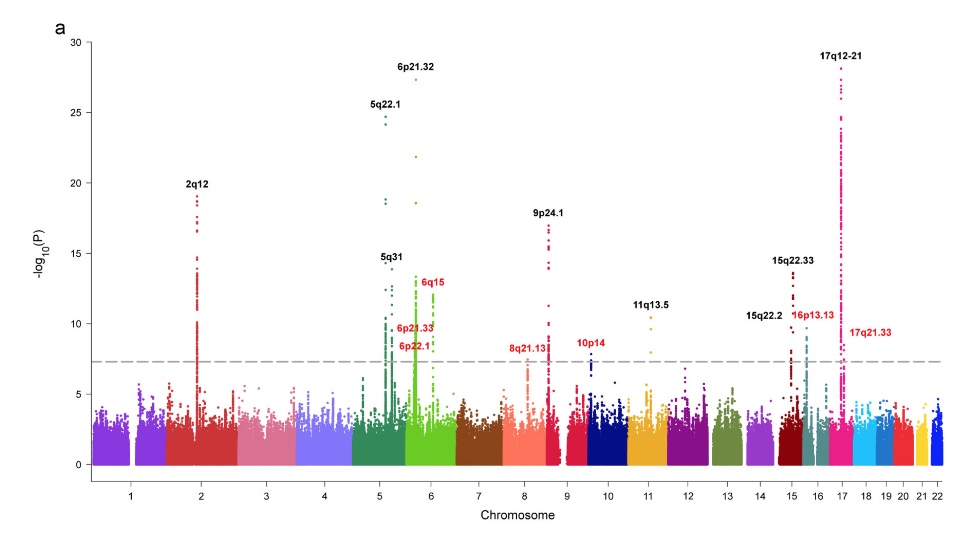
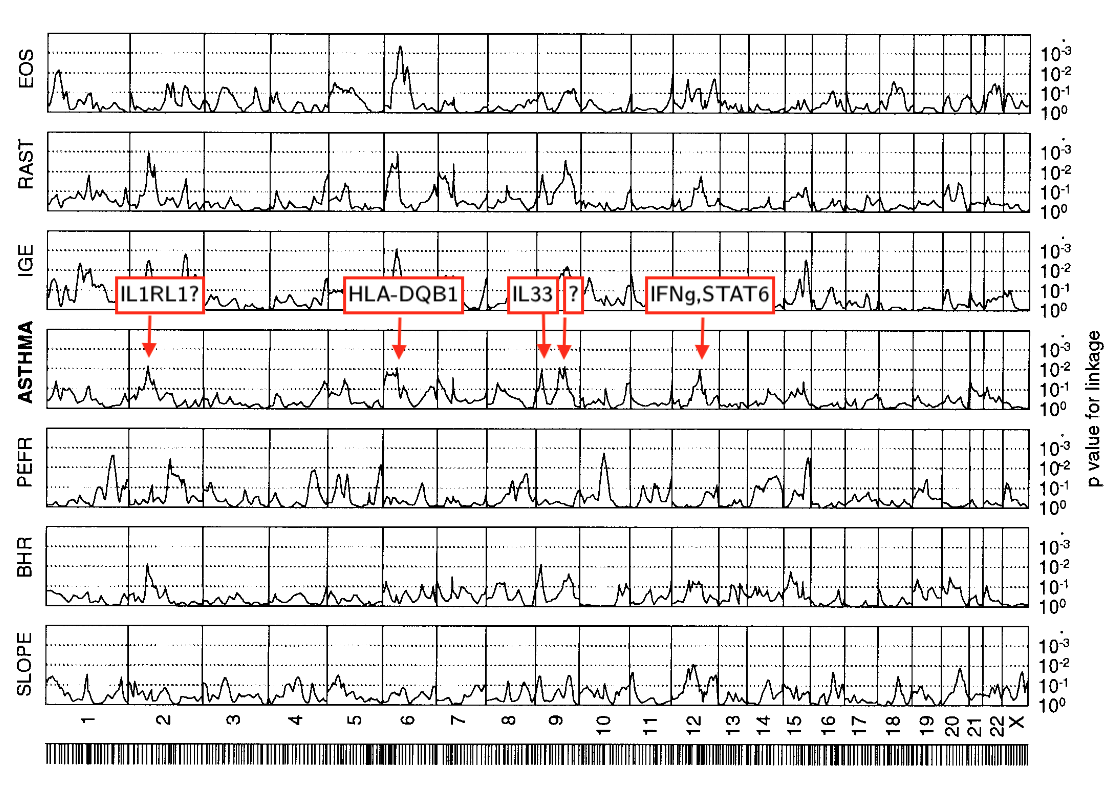
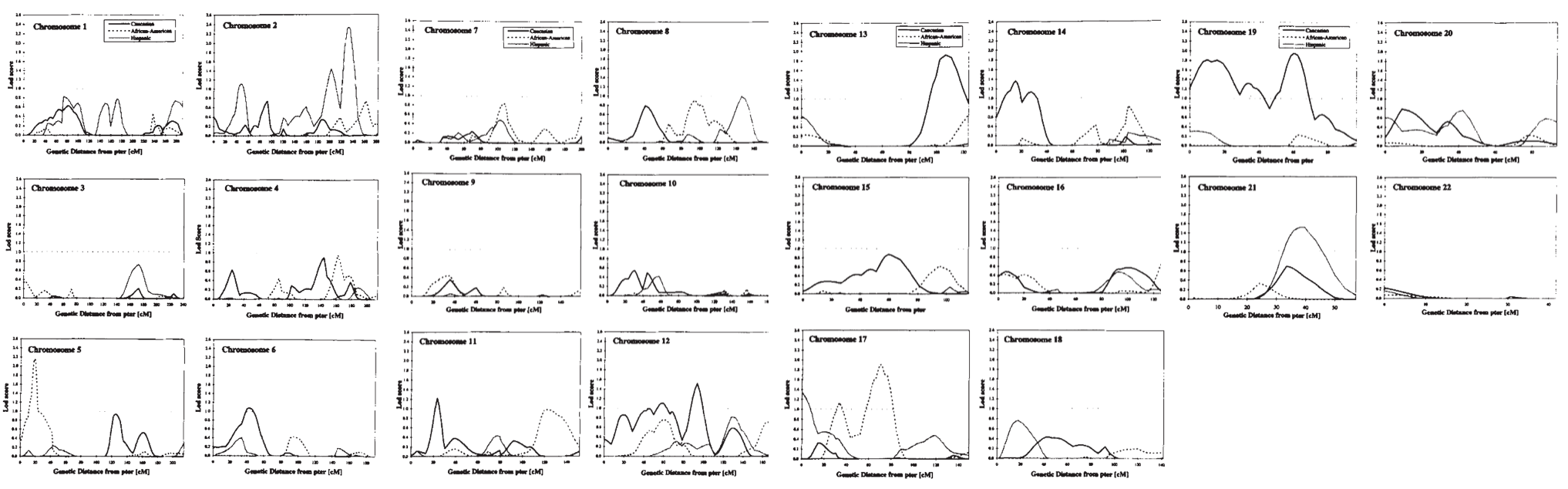
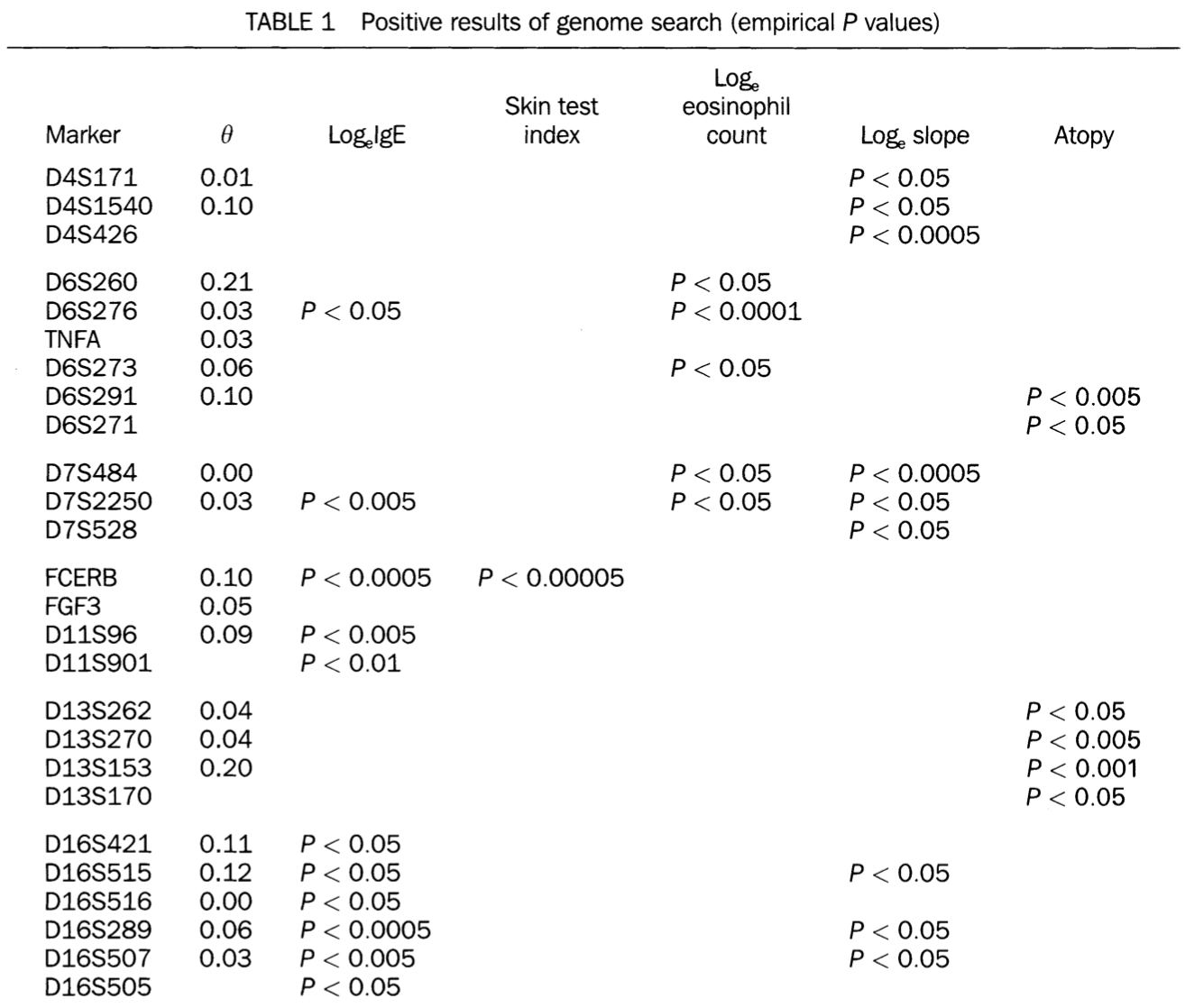
highly significant linkage in the genome scan (p=0.0001 for history of asthma and p=0.0009 for methacholine challenge) … at D11S907, a marker on the short arm of chromosome 11.
D11S907 or AFM109YA1 is a microsatellite marker located at 11p13 in a gene known as EHF (ETS homologous factor). There should be 2 genes in close proximity of the marker: ASTH1I and ASTH1J.
ASTH1I and ASTH1J were detected by exon trapping. ASTH1I exons detected a 2.8 kb mRNA expressed at high levels in trachea and prostate, and at lower levels in lung and kidney …
ASTH1J exons detected a 6.0 kb mRNA expressed at high levels in the trachea, prostate and pancreas and at lower levels in colon, small intestine, lung and stomach.
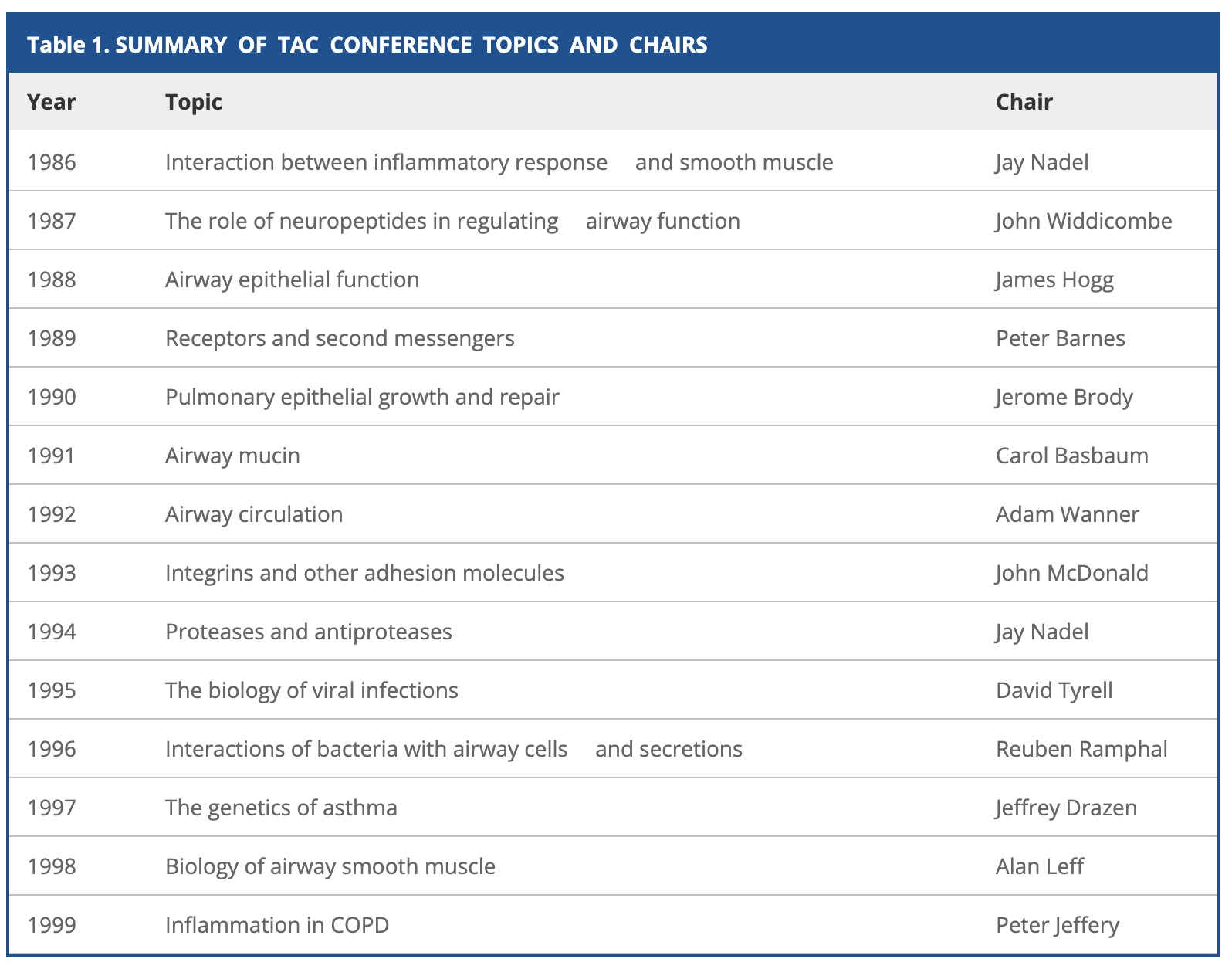
The conference series started in 1986 while the 12th conference was organized by Jeff Drazen with local support of Adam Wanner.
Sponsored by Boehringer Ingelheim, the participant list is the WHO is WHO in pulmonary genetics at that time: Bleecker, Meyers, Woolcock, Weiss, Burrows, Postma, Kauffmann, Dizier, LeSouef, Blumenthal, Banks-Schlegel, Slutsky, Zamel, Ober, Bousquet, Vercelli, Barnes, Adcock, Dahlen, Pauwels, Lewitt, Aron, Martinez, Cookson, Moffatt, Rosenwasser, Liggett, Rich, Papadopoulos, Levitt, Holgate, Elston, Morton and Marsh. Many of them do not live any more [1,2,3,4,5], some have made big careers [1,2], others have been fallen somewhat into disgrace [1,2,3] and many already retired. Continue reading 25 years Transatlantic Airway Conference 1997 on asthma genetics in Key Biscayne→
He basically killed one of my best papers by the Kruglyak-Lander rule of what is being “significant”. He started the stupid CRISPR origin discussion. But now he steps down
President Biden’s top science adviser Eric Lander resigned on Monday after an investigation revealed he violated the White House‘s workplace policy by mistreating staff members
Lander ‘retaliated against staff for speaking out and asking questions by calling them names, disparaging them, embarrassing them in front of their peers, laughing at them, shunning them, taking away their duties, and replacing them or driving them out of the agency. Numerous women have been left in tears, traumatized, and feeling vulnerable and isolated,’ Wallace told the outlet.
Statnews has a good commentary about “The fall of Eric Lander and the end of science’s “big ego’ era”
It’s not quite “big science,” which isn’t going anywhere. Call it “big ego.” In science, “big ego” isn’t exactly a new phenomenon. But in recent decades it grew with the emergence of researchers who could both handle the kind of gloves-off debate that can mark academic discourse and marshal vast resources to make certain types of scientific discoveries, like mapping genomes.
What is even more remarkable is the challenging keynote of the Xth World Congress of Psychiatric Genetics in The Palais des Congre ́s Brussels, Belgium October 9 –13, 2002 by Irving Gottesman [+2016], the father of epigenetics in psychiatry. He wrote there
We cannot escape the history of our field and are constantly guided today by the accumulation of facts with either positive or negative valences from our past. But when did the clock start—with the domestication of animals, with Galton’s musings and amoral passion for data collection about individual differences in behavior, or with the initially objective scientizing of Mendelism applied to schizophrenia but ending with a Nazi-tainted albatross around the neck of psychiatric genetics. In regard to the long quest for the distal and genetic (partial) causes of mental diseases, the conclusion that both genetic and environmental factors, none yet known in detail, provide the distal causes of mental disorders—that statement is too general to be of use to making further progress. What is needed is a confrontational approach based on evidence collected from competing ‘schools of thought’, and then reconciliation before some kind of omniscient and impartial Science Court.
I couldn’t agree more. What is needed is a confrontational approach based on evidence collected from competing ‘schools of thought’, and then reconciliation before some kind of omniscient and impartial Science Court.
Rewriting a local Python notebook now to Colabs (as I don’t have an eGPU and therefore also no CUDA support in macOS) I am now again restricted by a daily timeout…
This reminds me so much back to 1989/1990 when we programmed SAS on an IBM mainframe under VM/CMS. I usually went there early in the morning just to reserve my disk space :-) In the evening it was all gone just like Colabs nowadays…
***
* Auswertung des genetischen Einflusses...;
cms fi data disk dummy dummy e;
data a; set data.ges_crit(keep=
asthma frage f1ekzem f33heu f36ekzem);
***
BTW without an expensive GPU I had to use our HPC now. Will try a Jetson Nano or Google Coral in the near future as this seems to be more energy friendly.
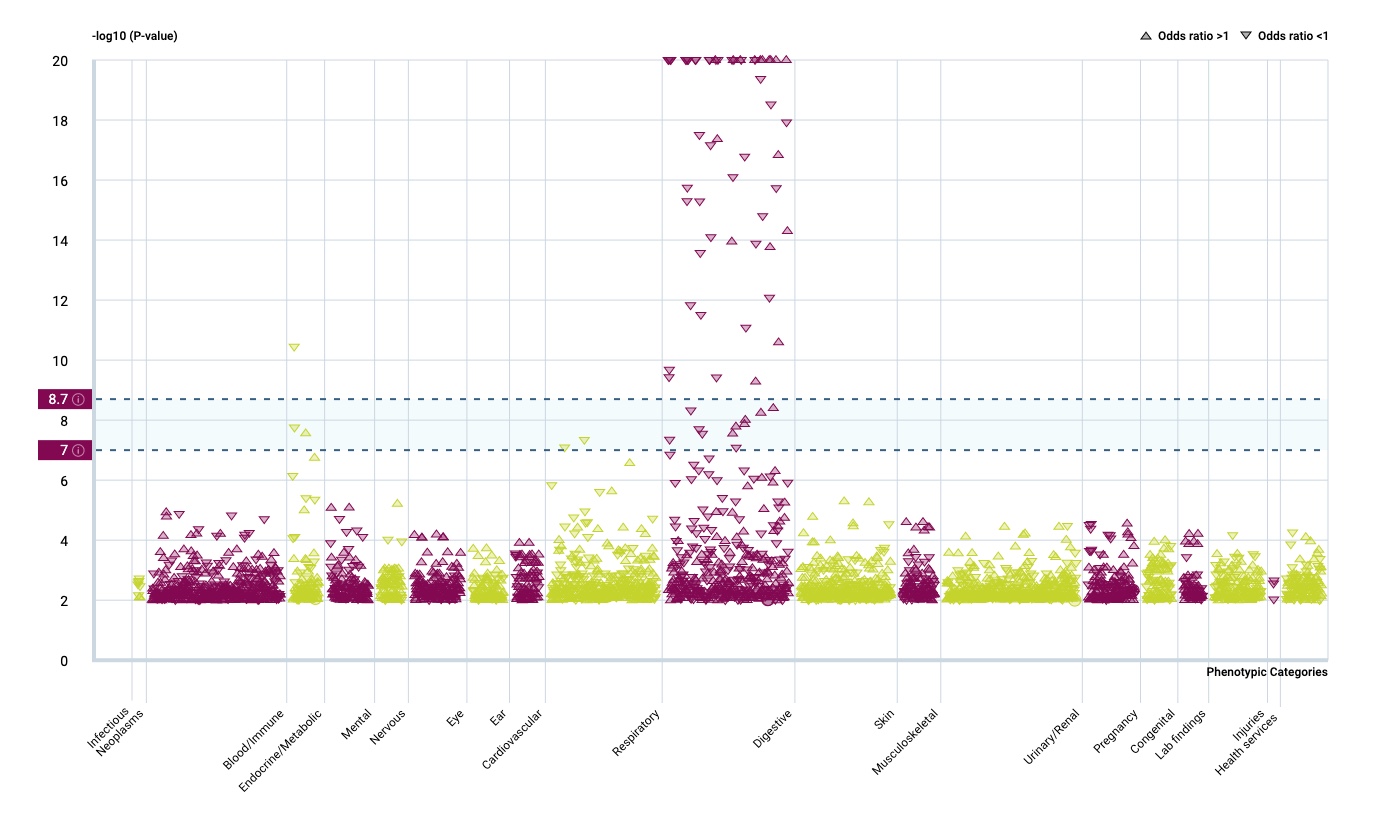
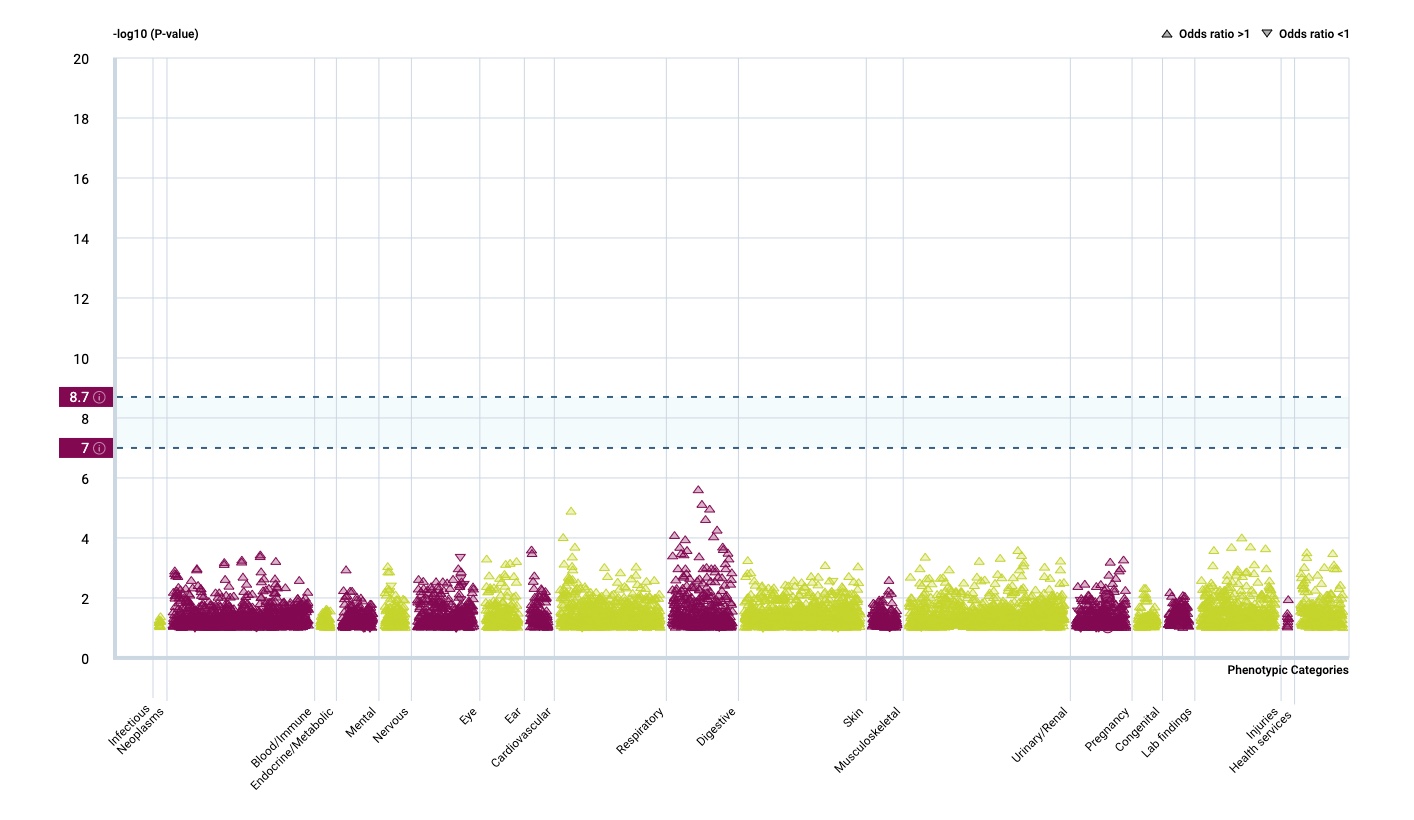
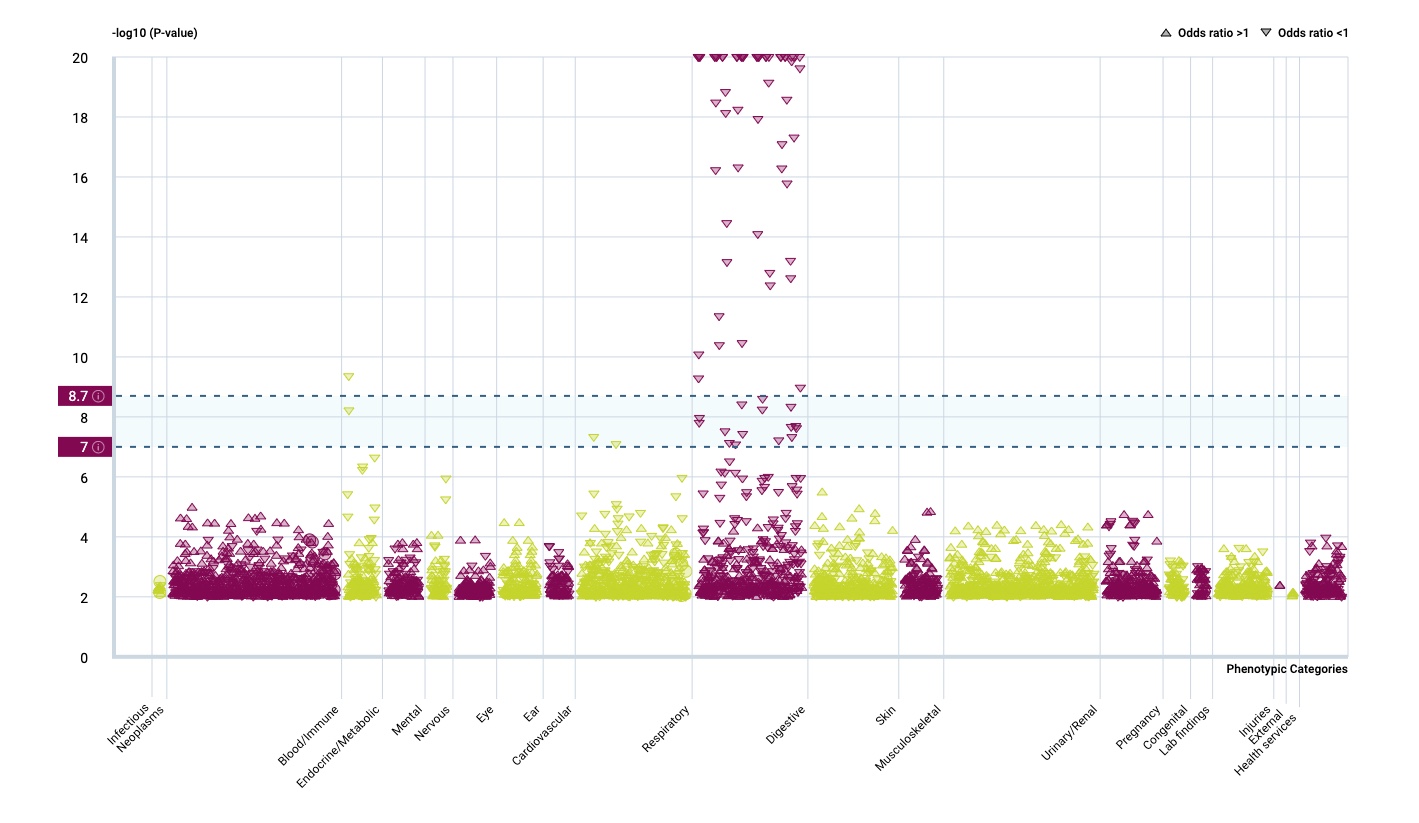
It seems that the respiratory tract isn’t so much influenced by rare gene variants but that there is a strong effect in the immune system.
And there is another interesting fact.
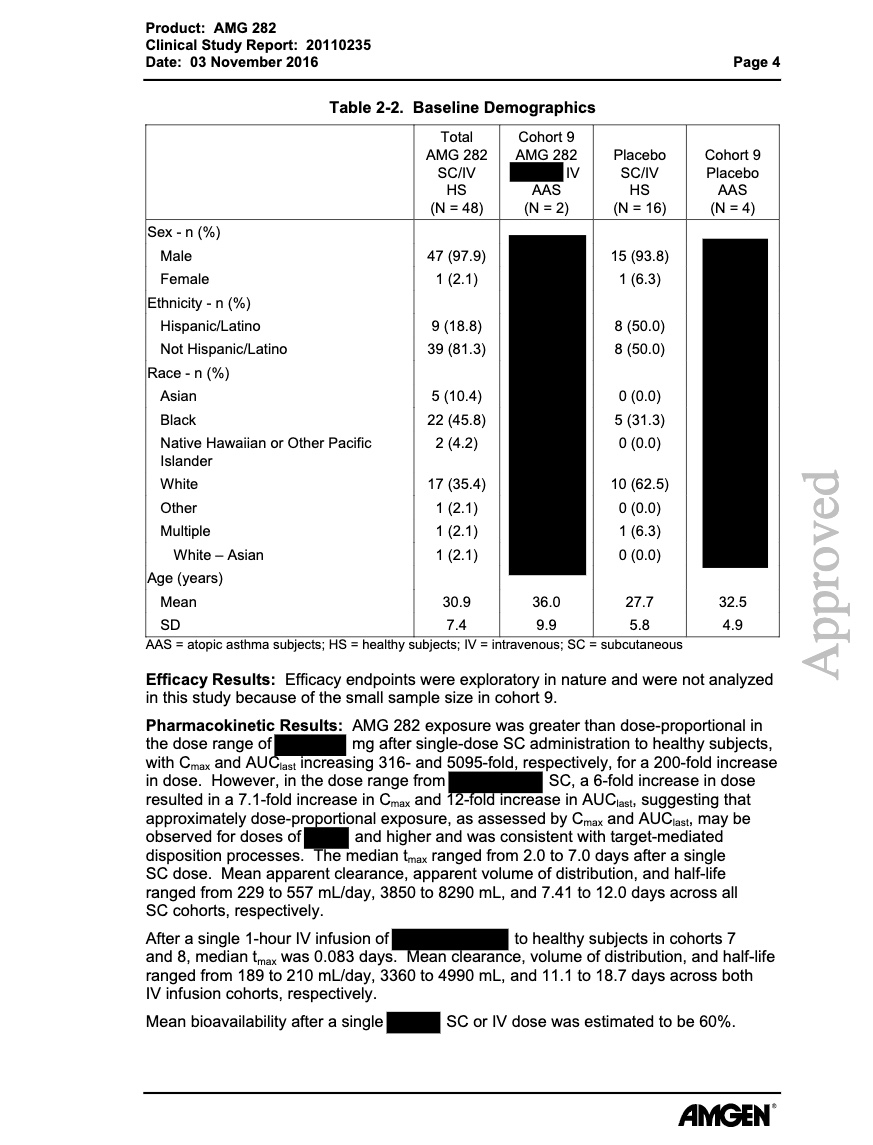
…Surveying the contribution of rare variants to the genetic architecture of human disease through exome sequencing of 177,882 UK Biobank participants …if we look at the …. European population who are carriers of a filaggrin (FLG) PTV, we find those carriers have significantly higher risk for well-known associations, such as dermatitis … and asthma … Concomitant increases in vitamin D levels suggest … increased sensitivity to ultraviolet B radiation.
So far, I have only assumed an asthma/allergy priming effect of oral vitamin D in the newborn gut. This paper now argues for an increased vitamin D sensitivity also in the skin of FLG dermatitis patients which is interesting given the largely contradictory data of serum vitamin D and atopic dermatitis. Maybe dermatologists should focus their research more on skin and local vitamin D turnover?
-II-
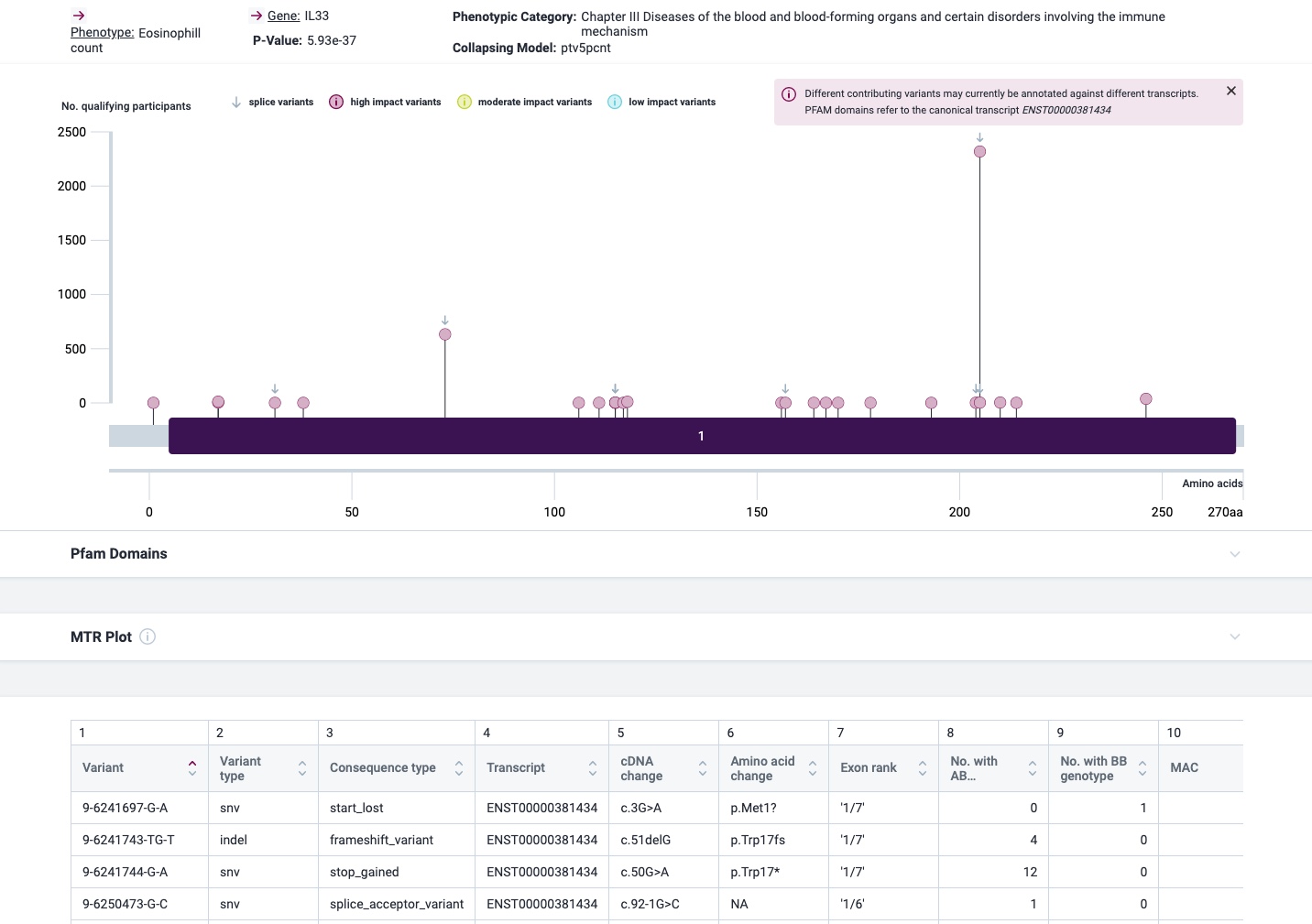
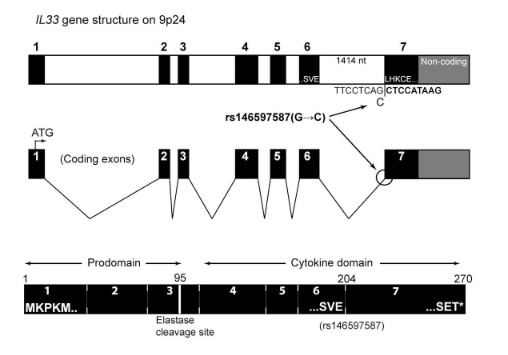
The most prominent IL33 variant carried by over 2,300 people is splice acceptor 9-6250473-G-C followed by 600+ individuals with splice donor 9-6250600-G-T.
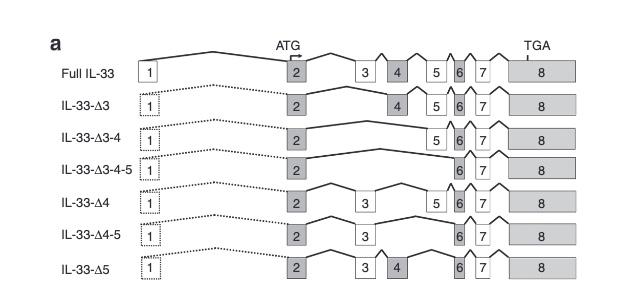
There are not too many carriers of this variant by the sheer amount of 177,882 participants. We nevertheless know already something about the seven IL33 splice variants since 2012.
Novel Splice Variants of IL-33: Differential Expression in Normal and Transformed Cells Journal of Investigative Dermatology (2012) 132, 2661–2664; doi:10.1038/jid.2012.180
Fig 3A Smith et al. A rare IL33 loss-of-function mutation reduces blood eosinophil counts and protects from asthma, PLoS Genetics 2017 – describes the splice site as NM_001199640:exon7:c.487-1G>C or rs146597587-C
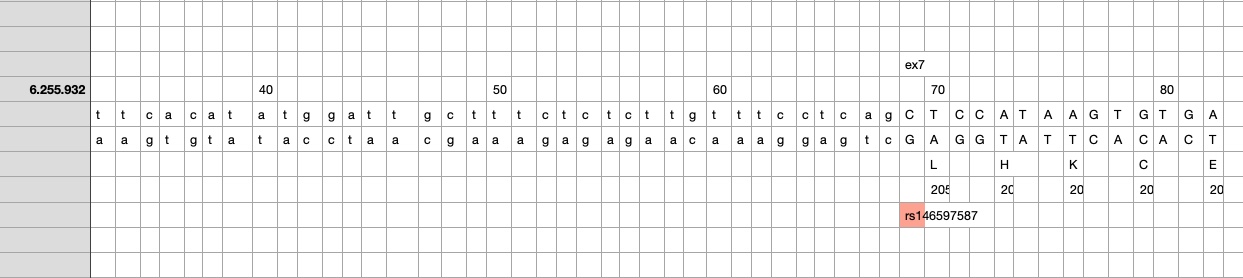
So I did a sequence match to compare the new finding with these older publications.
own sequence match exon7 using data from dbSNP, UCSC GoldenPath and Uniprot – reference is hg19
Indeed, the 2017 paper already described rs146597587 which is probably identical to the splice acceptor 9-6250473-G-C in Astra UK Phewas (genome positions do not match – I used hg19 while I don’t know the Astra reference) . Astra says also c.613-1G>C while rs146597587 is just upfront of my codon 205 (3*205=615) whatever that means.
The Astra UK Phewas at least confirms the Iceland paper above
rs146597587-C associates with lower eosinophil counts (ß= -0.21 SD, P = 2.5×10-16, N = 103,104), and reduced risk of asthma in Europeans (OR = 0.47; 95%CI: 0.32, 0.70, P = 1.8×10-4, N cases = 6,465, N controls = 302,977). Heterozygotes have about 40% lower total IL33 mRNA expression than non-carriers and allele-specific analysis based on RNA sequencing and phased genotypes shows that only 20% of the total expression is from the mutated chromosome. In half of those transcripts the mutation causes retention of the last intron, predicted to result in a premature stop codon that leads to truncation of 66 amino acids.
So it is basically a rediscovery meaning that we reached saturation.
A recent Nature study showed Ebola reactivation in a previously infected patient:
The 2021 lineage shows considerably lower divergence than would be expected during sustained human-to-human transmission, which suggests a persistent infection with reduced replication or a period of latency.
The most recent viral genome shared 10 substitutions that evolved during the previous epidemic making it unlikely that there was a new animal spillover event. I always wondered how second & third wave of COVID-19 started in Germany. Was it really a new spread or just a reactivation? A Frontiers review concluded from the existing literature that
our study, consisting more than a total of 113,715 patients, indicates that the RP-SARS-CoV-2 scenario occurs plausibly due to reactivation, reinfection, viral shedding, or testing errors.
So far, there are 240 documented COVID-19 cases of reinfection reported worldwide according to the reinfection tracker. In the case of Ebola there is a known viral persistence in semen while transmission through milk and cervicovaginal fluid is also possible (similar for COVID-19 although neuronal persistence seems to be more relevant). Maybe we need more immunological studies particular in long COVID if there is a continuous or intermittent antigenic stimulation due to persistence of an antigenic reservoir.
The fourth wave in Germany is caused by the delta variant, reactivation of alpha is certainly not a major factor. So we will only know in the next few years if reactivation is responsible for small regional outbreaks in unvaccinated communities.
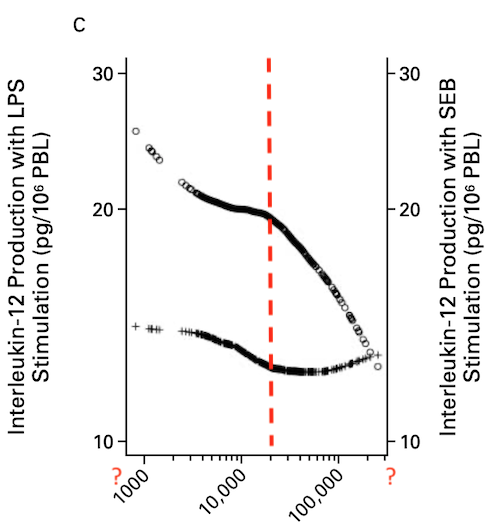
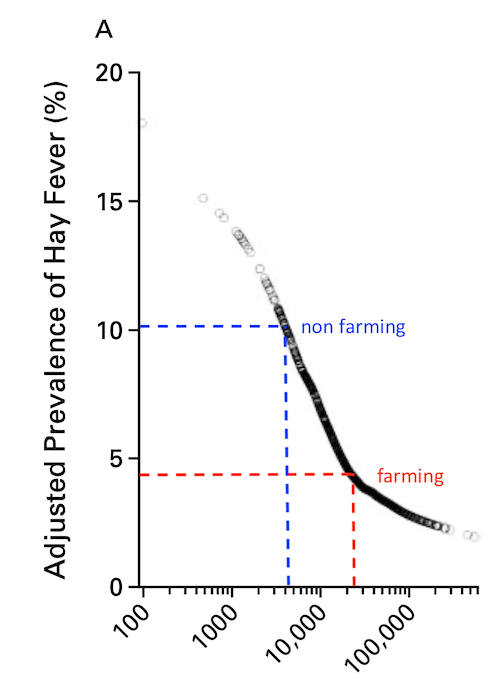
Is it justified to speak of a “protective” effect just by a negative association?
In addition to the problems with the math, I don’t get the point – farming should be leading to a generally reduced capacity for numerous pleiotropic cytokines?
There are even reports that LPS induces TH2 dependent senstization which is exactly the opposite of what this paper wants us to believe by some cryptic smoother applied to a heterogenous population.
On Feb 7,2020 I had the chance to hear a talk of Luke Jostins-Dean about Irritable Bowel Syndrome (IBS) and Inflammatory Bowel Disease (IBD). While IBS had a substantial overlap of polygeneic risk scores with psychological features, IBD did not. Sounds logical but does this prove anything?
Two new medRxiv preprints [Wendt and Marees ] throroughly examine also possible genetic correlates. Although I am quite sceptical that SES correlates should be tested at all (and also think that GWAS are not hypothesis free) here is the Marees explanation of the three possibilities we do have: PRS, MTAG, mtCOJO.
First, polygenic risk scoring (PRS) is a tempting approach; but PRS using mental health/disease to predict the same or different phenotypes from an independent dataset often explain very little variance in the outcome phenotype. PRS also cannot detect specific biology underlying each phenotype.
Second is multi-trait analysis of GWAS (MTAG), which jointly analyses GWAS summary statistics and adjusts per-SNP effect estimates and association p-values using the strength of the genetic correlation between phenotypes. Genetic correlations between EDU/SES and related phenotypes have, however, demonstrable biases from environmental confounders….
To disentangle the complex genetic overlaps between EDU/SES and mental health, we therefore used multi-trait conditioning and joint analysis (mtCOJO), which generates conditioned GWAS summary statistics for each phenotype of interest after correcting for the per-SNP effects of another phenotype). The mtCOJO approach is not based on genetic correlation; it is based on the causal relationship between trait pairs inferred by Mendelian randomization (MR).
So, more than 2 decades ago we found hits on chromosome 2, 6, 9, 12 (missing chr17q21 where our marker coverage wasn’t probably good enough). It seems that this was the first identification of the IL33 region although IL33 was described only 7 years later. Remarkably, this result was possible with just 415 individuals instead of 500,000 individuals nowadays (see also the asthma genetics timeline).
1 x ejaculation expells 250,000,000 sperm at a speed of 500 cm/s. Each sperm contains 3,088,000,000 base pairs = bits~ 368 megabyte of genomic DNA
Each base pair takes 2 bits (you can use 00, 01, 10, and 11 for T, G, C and A). … And remember, you have to go from bits to bytes to get to an answer in megabytes. A bit is just a single unit of digital information, but a byte is a sequence of bits (usually 8). And because computers work in binary math, 1 kilobyte = 1024 (i.e. 2 x 2 x 2 x 2 x 2 x 2 x 2 x 2 x 2 x 2 = 1024). 1 gigabyte = 1024 megabytes = 1048576 kilobytes = 1073741824 bytes. So you take the 3,088,000,000 bits and divide it by 8 to get 750,000,000 bytes. Divide that by 1024 and you get 376,953 kilobytes. Divide it by 1024 once more and you’re left with 368 megabytes.
Add 30,000 CpG islands x 8 bytes ~ 0,2 Megabyte
Also add 75 mitochondria x 16,569,000 base pairs ~ 148 Megabyte
So in total 516,2 Megabyte per sperm
the practice of utilizing the DNA of an individual to predict disease has been judged to provide little to no useful information.
but they nevertheless the group tried to rescue the concept by combining clinical risk factors plus low/medium/high polygenic risk. As a pragmatic approach this may work for some diseases but it will not explain any genetic pathhway.
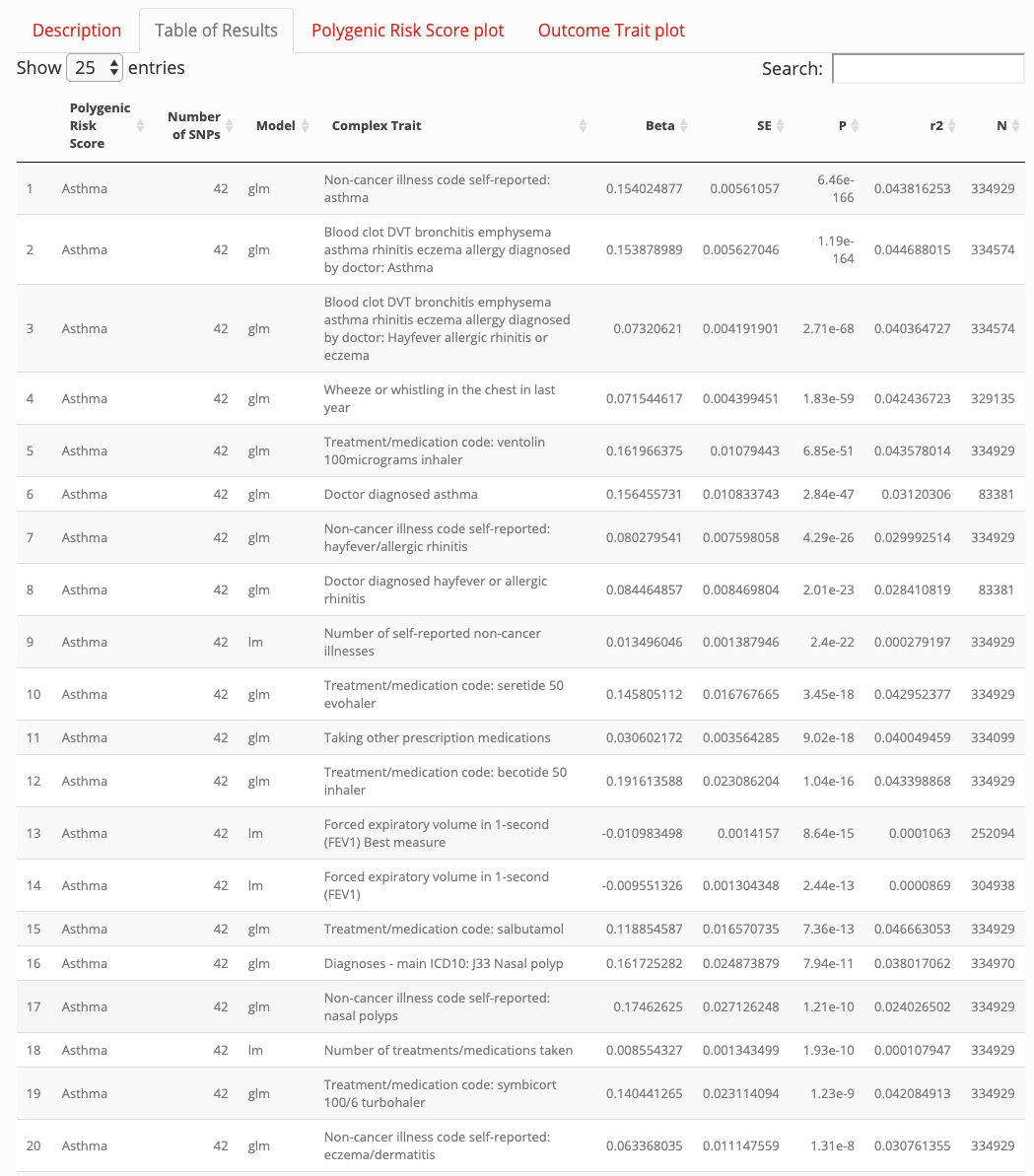
More recently Richardson et al. tried another approach using the UK Biobank data where they analysed 162 PRS and 551 heritable traits from 334,398 individuals. Using their web application for my search term “asthma” I did not find anything useful that hasn’t been known for ages.
Jan 15, 2020 http://mrcieu.mrsoftware.org/PRS_atlas
So lets have a look at an example published last November in Cell “Screening Human Embryos for Polygenic Traits Has Limited Utility”. The outcome is sobering: If an IVF embryo would be profiled with polygenic scores for traits such as height or IQ
the top-scoring embryo is expected to be about 2.5 cm or about 2.5 IQ points above the average. The adult trait value of the top-scoring embryo would remain widely distributed.
I wouldn’t have even run this analysis for being pointless.