Sydney Brenner has written for many years a column in Current Biology called Loose Ends (Nature called him a man who talks a lot which is certainly unfair or at least not empirically proven – we are talking between 20,000 and 40,000 words every day). Loose ends seem to have influenced a whole generation of biologists – kitsch biology included. Yea, yea.
Tag Archives: Genetics + Biology
Another layer of complexity in gene regulation
Yesterday evening I attended an excellent presentation by Nikolaus Rajewksy about microRNAs, small noncoding RNAs that are thought to have a role in posttranscriptional regulation. Nikolaus just moved 3 months ago from New York to follow Jens Reich at MDC in Berlin). Basically, he talked about his recent “l(ou)sy” paper and the “SNP” paper after giving a rather detailed history about the development of the field. It started in 1950 with Jacob and Monod, 1960 Britten and Davidson, 1970 Haywood (who even quit science after being dissappointed), finally to 1990 when the Ambros and Ruvkun labs discovered nematode microRNAs. Current research is mainly done in the Tuschl, Batel, Cohen, Lander and Rajewsky labs who produce the bulk of the 800 papers or so published in 2006.
Approximately 30% of genes are influenced by microRNAs, the total number of microRNA sites is under heavy debate (~22,000) as well as the number of human microRNAs (328); each microRNA regulates ~200 genes. Unfortunately there is still no highthroughput technique to detect targets. There is also no good prediction by free energy and even mismatches in the 5 prime of mRNA are possible (individual predictions can be obtained at Pictar that uses a hidden Markov model).
If I understood that correctly, miRNA are the feedback mechanism on RNA level (with transcription factors at the DNA level). He mentioned 3 classes known so far in humans: oncomiRNA, miRNA 375 myotrophin, and miRNA 122 acting on cholesterol (quite interesting as being described recently in the NEJM. The experimental knockdown of liver specific mouse microRNA shows ~300 up- and ~300 down regulated genes. Upregulated genes have in approximately 50% of cases one miRNA nucleus, downregulated ones have even less than average binding sites. There is no overrepresented GO category in upregulated genes but cholesterol is highly significant in downregulated genes whatever that means. Action of miRNA seem to heavily context dependent giving us many more questions than answers. Yea, yea.
All roads to NLM
This is not just an addendum to my previous post free-for-all or to number-cruncher: the 12 Dec NIH press release links to a new and exciting database
NIH Launches dbGaP, a Database of Genome Wide Association Studies
The National Library of Medicine (NLM), part of the National Institutes of Health (NIH), announces the introduction of dbGaP, a new database designed to archive and distribute data from genome wide association (GWA) studies. GWA studies explore the association between specific genes (genotype information) and observable traits, such as blood pressure and weight, or the presence or absence of a disease or condition (phenotype information).
Addendum
29-5-07 dbGaP suffers from some broken links but content improves!
3D LD
While waiting for genomewide SNP data to be re-partioned into LD blocks I found this page with some neat progamming tricks. It is part of the dissertation of Ben Fry / MIT about computational information design. Page 74 ff has a history of redesigning the widely used haploview pogram.
The design of these diagrams was first developed manually to work out
the details, but in interest of seeing them implemented, it was clear that
HaploView needed to be modified directly in order to demonstrate the
improvements in practice. Images of the redesigned version are seen
on this page and the page following. The redesigned version was even-
tually used as the base for a subsequence ‘version 2.0’ of the program,
which has since been released to the public and is distributed as one of
the analysis tools for the HapMap [www.hapmap.org] project.
How can we know?
A recent paper in Nature reported
Tissue samples were obtained from one of the following sources: Asterand, Pathlore, Tissue Transformation Technologies, Northwest Andrology, National Disease Research Interchange and Biocat. Only anonymized samples were used, and ethical approval was obtained for the study from Ärztekammer Berlin and the Cambridge Local Research Ethics Committee. […] Human primary cells were obtained from Cascade Biologics, Cell Applications, Analytical Biological Services, Cambrex Bio Science and the Deutsches Institut für Zell- und Gewebeersatz.
How did “Ärztekammer Berlin” or “Cambridge LREC” evaluate ethical performance of these companies? Or did anonymity automatically guarantee ethical research? Or is it just a formal requirement to mention ethics? Or …?
Evolution in fast motion
Nature genetics as an advance online publication about comparative genome sequencing of E. coli where 13 de novo mutations in 5 strains were monitored over 44 d (or ~660 generations). It is a great study – not only because the author list includes one of my previous coauthors – but for giving a first insight about development of a mutation and fixing its allele frequency. Unfortunately, there is no flowchart and the methods are somewhat vague, what has been sequenced (or resequenced) in which strain at what time . In other words who are the winners? Did they manage that by their own strength or with a little help of some friends? Why rises the allele frequency always to 100% and what about some discrepancy of allele frequency and fitness? We will hopefully see more of these studies, yea, yea.
Pathway to nowhere
I love pathway diagrams since I mounted the famous Biochemical Pathways of Boehringer in my bachelor flat. As far as complex disease genetics is concerned with many disease genes, an integration into a pathway context becomes critical. There are many attempts to extract this information from the literature and many companies that offer highly curated information (Biomax, Ariadne Genomics, Genomatix to name a few). Academia relies mainly on KEGG, the Kyoto encyclopedia of genes and genomes or Biocarta. Last week another pathway server appeared that is curated by the NCI and Nature magazine. Let’s have a look – I am currently working on Affymetrix 500K SNP annotation, yea, yea.
How to detect your own CNVs
How to detect copy number variation (CNV) in your own genotype chip data, can be found in a companion paper of the recent Nature publication.
In the previous Nature paper the authors explained their algorithm to be based on k-means and PAM (partitioning around medoid) clustering, but it seems quite different. They call genotypes with DM (which seems to be already obsolete by the BRLMM, see a comparison at Broad and the AFFX whitepaper), then adjust heterocygote ratios by Gaussian mixture clustering, normalize and reduce noise before! merging NspI and StyI arrays. The software is at Genome Science, Tokyo. Yea, yea.
On mice and men with asthma
A new series of pro- and con editorials in the Am J Res Crit Care Med discusses the question why in some instances mouse models have “misdirected resources and thinking”. You may have noticed that I have only rarely used animals for research; the authors of this editorial have collected empirical data on the exploding use of murine models. Despite their attractiveness from a technological point, they are often useless because
- mice do not have asthma as even the most hyperresponsive strain does not show spontaneous symptoms
- mice do not have allergy – although sensitization can be manipulated by high intraperitoneal allergen/adjuvant injection, this does not involve immediate and late airway obstruction.
- immune reaction in mice is quite different – the interfering of some substances like vitamin D cannot be reliable tested, there is no pure Th1 and Th2 reaction in human and less stronger IL-13 response
- mice typically can not be challenged with the complex (and interacting) human exposure – oxidant stress, viral infection, obesity, diet, smoke, pollutants, ….
- time course is difficult to mimicking in the mouse, there is no longterm model
- structure of mouse airways is different – there are fewer airway generations, much less hypertrophy of smooth muscle
- inflammation in mouse is parenchymal rather than restricted
- humans are outbred, mice are inbred
- early microbial environment is different
- many promising interventions of mice pathways failed in humans (VLA-4, IL4, IL5, bradykinine, PAF,…)
I am sure there are even more arguments – I suggest that the authors deserve the Felix-Wankel price.
Addendum
15 Dec 2006: The BMJ has 6 more examples about the discordance between animal and human studies: steroids in acute head injury, antifibrinolytics in haemorrhage, thrombolysis or tirilazad treatment in acure ischaemic stroke, antenatal steroids to prevent RDS and biphosphonate to treat osteoporosis.
19 Dec 2006: Another pitfall paper
31 Dec 2006: A blog on animal welfare
25 Apr 2007: Call for better mouse models
15 Jul 2018: Of Mice, Dirty Mice, and Men: Using Mice to Understand Human Immunology
7 Jan 2023: A review concluding that The vitamin D system in humans and mice: Similar but not the same
Why dog and cat can´t marry
or in more scientific terms: Why are F1 hybrids so often sterile or lethal? The Dobzhansky-Muller theory says that there is an incompatibility between genes with reduced fitness that have diverged between species. So far nobody has ever observed a D-M gene but a new Science paper describes two genes that separate D. simulans and D. melanogaster: lhr (lethal hybrid rescue) and hmr (hybrid male rescue). Yea, Yea.
For the first time two human genomes compared
Another “first discovery” in this nature genetics preprint although the analysis could have already been done some years earlier. The CNV specialists from Toronto now compare the Human Genome Project sequence with the Celera sequence – the gap between the two compilations was obviously bigger than the intra-sequence gaps. Of course both sequences are still mosaics from several individuals but the analysis nicely exemplifies how difficult it will be to compare the genome of two different human beings.
The authors employ a whole battery of alignment tools BLAT, MEGABLAST, GCA and A2Amapper. Of course results depend on the strategy, definition and implementation. As show by FISH analysis most of the discrepancies are true and can be classified into a few categories – insertions or deletions if seen from the second genome (has somebody ever thought about a minimal human genome?), mismatches and inversions. We are getting here a preview of the diagnostic workup in a patient in 2026. This blog contains forward looking statements while the responsibility rests solely with the reader. Yea, yea.
New R packages for SNP studies
An utter refutation
I am slow in commenting on a paper that has already been published earlier this year – Joe Terwillingers vivid refutation of the fundamental theorem of the hapmap proponents that
if a marker is in tight LD with a polymorphism that directly impacts disease risk, as measured by the metric r^2, then one would be able to detect an association between the marker and disease with sample size that was increased by a factor of 1/r^2 over that needed to detect the effect of the functional variant directly
I cannot comment on the statistical proof but fear from my recent experience with Crohn and asthma tags that he may be right with his assumption: Even marker in high LD with the functional variant may not show any association at all. These may be bad news for all those currently running large screening programs with hapmap based variants believing that P(A|BC)=P(A|Bc)=P(A|B), yea, yea.
Addendum
Tag SNPs also do not work with CNVs
Who will survive?
When looking at gene variants in a population we may forget that even having a perfect sampling scheme this will not be an unbiased view of the human genome. Earlier studies suggested that up to 75% of conceptions are lost during early development; a further indicator of an biased view are unexplained cases of departure from Hardy-Weinberg equilibrium.
Selective survival during early pregnancy is still a terra incognita and except of studies in the Hutterites I am not aware of any (modern) study that looked at selective survival.
A study of Grant Montgomery now shows fresh data on genomewide allele sharing in 1,592 DZ twins from Australia and 336 DZ pairs from the Netherlands.
It is somewhat disappointing that there is no excess allele sharing in the HLA region nor somewhere else in the genome. Maybe further studies can do that a high resolution than with just 359 microsatellite marker?
Escaping from a swamp
The November AJHG has an excellent re-analysis of the dysbindin-schizophrenia association using new methodology that surpasses all previous meta-analysis techniques. As the single SNP association results from the previous 6 studies cannot be directly compared, they construct a European super-hap map from all tag SNPs in that region, place them in a phylogenetic tree before finally mapping all single associations on these haplotypes. Their Fig.1B show the main results; as the circles in Fig.1B are somewhat confusing, I have withdrawn their results – adding the haplotype frequencies and ordering the studies by year of publication.
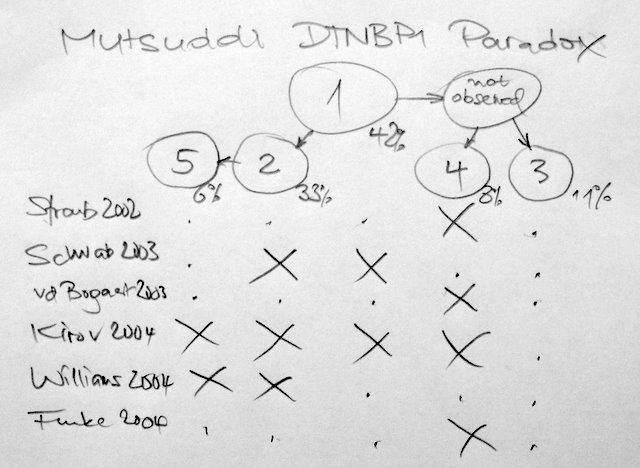
We may think of a triple-blind study – neither patients, nor PIs, nor we did know anything before. The results are alarming. I do not understand how the Kirov set could have included all haplotypes and why the Schwab/Williams set is in opposition to the Straub/Bogaert/Funke set.
What could have gone wrong? The authors of the current re-analysis believe that population differences are an unlikely reason for the inconsistency as the allele frequencies match between studies. Good news that genotyping errors may be largely excluded.
Unfortunately the authors remain vague why there is no common causal variant. Have there been different sampling schemes, different diagnostic thresholds, different environmental exposures in the previous studies? Is dysbindin at all a schizophrenia gene, or only under a certain genetic background? It seems possible that studies of one branch are false positives. Or is the haplotype reconstruction in the re-analysis erroneous for whatever reasons?
Von Münchhausen is well know for escaping from a swamp by pulling himself up by his own hair. I would like I could do that too.