While optimizing the analysis strategy for a 500,000 SNP Affymetrix array set, I found 6 autosomal SNPs that show highly significant sex-dependent allele differences: rs2809868, rs4862188, rs2880301, rs3883011, rs3883013 and rs3883014.
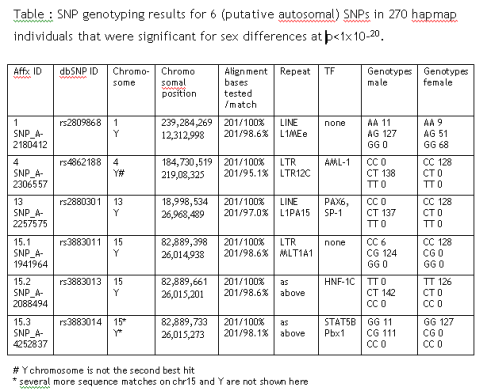
Sure, there could be autosomal marker that influences male/female outcome but there is a more likely explanation: All SNPs have paralogue sequence stretches on the Y chromosome that are co-amplified during PCR. From the initial genotyping results it is most likely that only the Y chromosomal stretch is being mutated in SNP 4, 13 and 15.2.
These SNPs are perfect sex marker, as they include an autosomal control allele (in comparison to pure Y markers like SNPs in SRY). They are always unambiguous (in contrast to pure X marker where only heterocygotes are informative).
They even offer advantage to commercial STR kits of the Amelogenin/Amely gene situated (in the Y parautosomal region) as they would not be affected by excess homologous X chromosomal material as often found in forensic situations. In addition, they might overcome some other weakness of the Amelogenin test where a second assay is usually recommended.
If you will ever see a case-control study that is highlighting any of these SNPs, you can be sure that this study had a distorted male-female ratio between case and controls.