The AJHG preprint server has an important paper about the effect of rare missense alleles. By combining information from HGMD, human – chimpanzee divergence and 4 other datasets (NIEHS-EGP, Seattle SNPs, JSNP and a resequencing approach of 58 genes in >1,500 chromosomes) they attack the Chakravarti hypothesis of “common diseases – commmon variants”
It remains uncertain why such polymorphisms can persist without being eliminated by purifying selection. Currently, two major lines of reasoning exist that explain this apparent paradox. The first considers various complex evolutionary scenarios and treats positive or balancing selection as a major force that can drive medically detrimental mutations to high frequencies. The second line of reasoning postulates a high mutation rate as a major factor that determines the cumulative frequency of detrimental polymorphisms in the population.
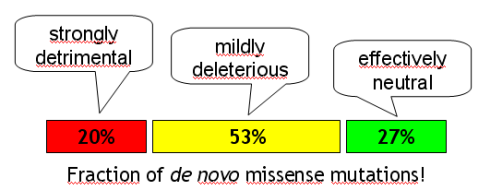
Anyway, here is the main outcome
CC-BY-NC Science Surf , accessed 23.07.2026