A quick update of my DNA pooling studies: The Affymetrix Genotyping Console 1.0 now includes a parameter CONTRAST Continue reading And now all together
CC-BY-NC Science Surf , accessed 26.07.2026
A quick update of my DNA pooling studies: The Affymetrix Genotyping Console 1.0 now includes a parameter CONTRAST Continue reading And now all together
When starting again analysis of pooled DNA samples – this time on the Affymetrix platform – I discovered not only the much improved “Genotyping Console” but also that my earlier Genepool binaries do not work anymore under VMWARE/Ubuntu. I therefore tried Continue reading My linux box
While optimizing the analysis strategy for a 500,000 SNP Affymetrix array set, I found 6 autosomal SNPs that show highly significant sex-dependent allele differences: rs2809868, rs4862188, rs2880301, rs3883011, rs3883013 and rs3883014.
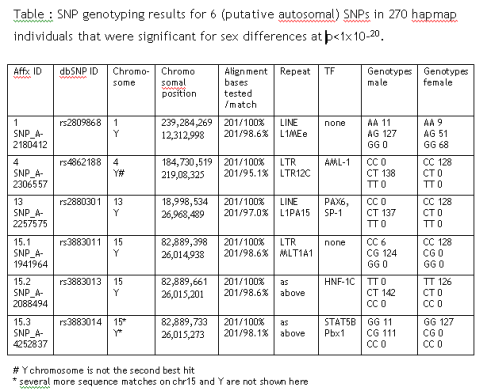
Sure, there could be autosomal marker that influences male/female outcome but there is a more likely explanation: All SNPs have paralogue sequence stretches on the Y chromosome that are co-amplified during PCR. From the initial genotyping results it is most likely that only the Y chromosomal stretch is being mutated in SNP 4, 13 and 15.2.
These SNPs are perfect sex marker, as they include an autosomal control allele (in comparison to pure Y markers like SNPs in SRY). They are always unambiguous (in contrast to pure X marker where only heterocygotes are informative).
They even offer advantage to commercial STR kits of the Amelogenin/Amely gene situated (in the Y parautosomal region) as they would not be affected by excess homologous X chromosomal material as often found in forensic situations. In addition, they might overcome some other weakness of the Amelogenin test where a second assay is usually recommended.
If you will ever see a case-control study that is highlighting any of these SNPs, you can be sure that this study had a distorted male-female ratio between case and controls.
I truly liked the recent Sjoblom study while a new Science letter now raises heavy criticism:
… put into stark reality the challenges facing the Human Cancer Genome Project (HCGP). One wonders about the merits of such high-cost, low-efficiency, and ultimately descriptive-type “brute force” studies. Although previously unknown mutated genes were unearthed, the functional consequences of most of these and their actual role in tumorigenesis are unknown, and even with that knowledge we are a long way from identifying new therapeutic targets.
This seems to be the open wound of modern biology: all these high throughput driven genotyping / expression profiling / metabolome scanning approaches are mainly money & impact & activity driven – parameter or hypothesis-free has become a fashionable buzz phrase while only a few years ago it would have been an affront to every serious researcher.
Funny to see also the new Nature initiative opentextmining.org as nobody wants to read the results of these studies. So at least computers should be able to do that. Fail better…
Similar criticism of the Neanderthal studies but a different argument
However, although such comparisons are of interest, it is not the static genome but rather the dynamic proteome that determines the phenotype of an organism. Salient examples include the caterpillar and the tadpole, which share
genomes with the butterfly and frog, respectively, but which have very different proteomes making them into very different organisms.
Thus, rather than performing untargeted comparisons of sizable genomes, we suggest that it might be more useful to address this question using a standard hypothesis-driven approach.
My interest in DNA pooling was always strong; we have developed methods doing this on the mass spec platform and applied it to the HLA region. I had, however, doubts if testing pools by less accurate methods like chip hybridization will work. The January issue of the AJHG now has a fascinating article how pooling may even work on the Affymetrix platform. Yea, yea.
DNA pooling can be even used in family context, see Wen Chung Lee in Cancer Epidemiology 2005 or Neil Risch in Genome Research 1998.
Finally, linkage and association data can be used together after downloading new software using genotype inference.
It reduces the number of genotyping reactions and increases the power of genome-wide association studies. Our method combines sparse marker data from a linkage scan and high-resolution SNP genotypes for several individuals to infer genotypes for related individuals.
Sure, we
but this seems to be the best recycling for our old fashioned linkage data. Yea, yea.