How to recognize photoshopped pictures? This will be a routine task for future editorial process (BTW I already recognized a faked gel gel picture where the edges and density of the bands looked somewhat artifical). However, with the ever increasing technical capacities we probably need non-destroyable, watermarked pictures from professional scanning and digitizing equipment.
In the meantime, check Wikipedia and the links there. I believe that the majority of the faked pictures could have been detected by splitting up color channels and looking at non-continous transitions of hue (“Farbton”), saturation (“Sättigung”) and brightness (“Helligkeit”) or grey value. This will even work with scanned figures although I would recommed to check original computer files (that may always be electronically stamped by previous publishers). Don´t miss the website of the mp3 developers).
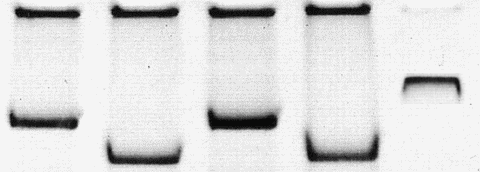

Addendum
Here is another examples how to recognize photoshop spoof:
set the hue to a low setting, the saturation to a higher setting, and mess with the light and look for blotches of color that don’t follow the rest of the image
22-2-07: The JBC has now adopted an explicit policy
“No specific feature within an image may be enhanced, obscured, moved, removed, or introduced. The groupings of images from different parts of the same gel, or from different gels, fields or exposures must be made explicit by the arrangement of the figure (e.g. using dividing lines) and in the text of the figure legend. Adjustments of brightness, contrast, or color balance are acceptable if and as long as they do not obscure or eliminate any information present in the original. Nonlinear adjustments (e.g. changes to gamma settings) must be disclosed in the figure legend.
11-9-07 Hamin Farid has developed tools to detect digital tampering.