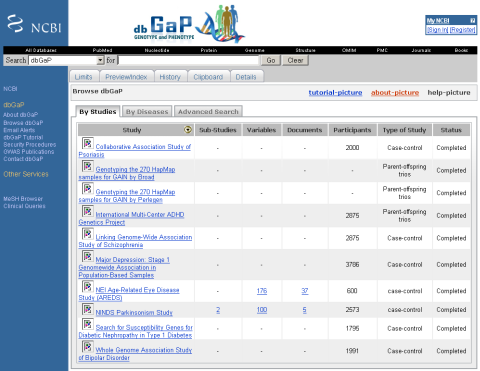
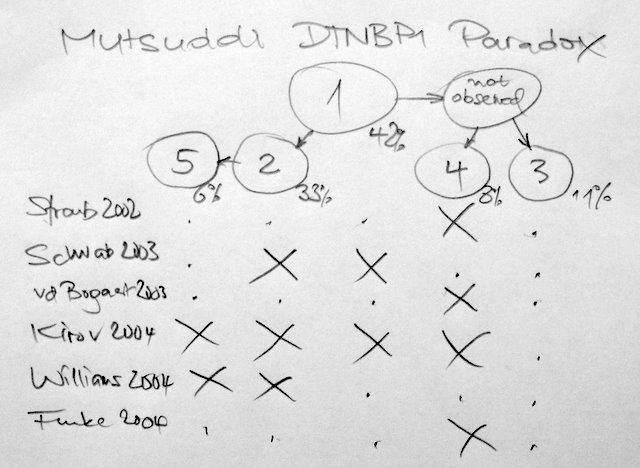
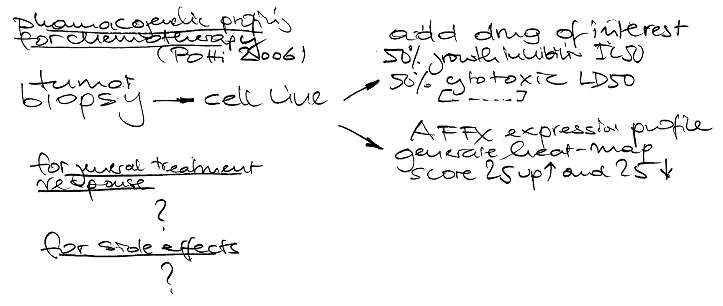
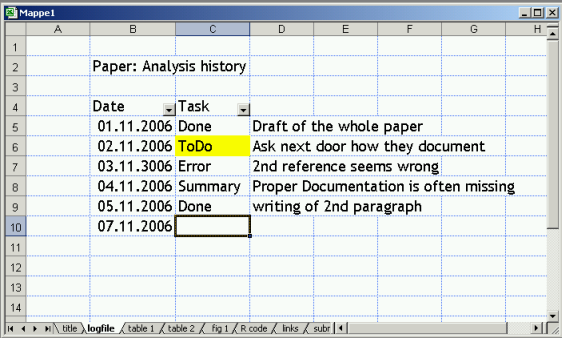
Looking again at human variation it seems that my recent estimate of 99,9% sequence identity is wrong as shown in an nature editorial yesterday and of course the new paper with the first copy number map of the human genome
3,080 million ‘letters’ of DNA in the human genome
22,205 genes, by one recent estimate
10 million single-letter changes (SNPs) â€"
that’s only 0.3% of the genome
1,447 copy-number variants (CNV),
covering a surprisingly large 12% of the genome
About 99.5% similarity between two random people’s DNA
I am organizing my literature in folders where the CNV section is still very thin but labelled as high priority – this seems to be adequate as the new study shows that the CNV emcompass hundreds of genes and functional elements.
Notably, the CNVRs encompassed more nucleotide content per genome than SNPs, underscoring the importance of CNV in genetic diversity and evolution
The Wellcome Trust has a nice website about copy number variants. If you want to read more, you will find information about the methods (array-based comparative genome hybridisation, cytogenetics, population genetics, comparative genomics and bioinformatics) as well as the questions that drive CNV research.
Again it seems that disease genetics is not only about stupid nucleotide polymorphisms (SNP), it is a whole bunch of chromosome aberration, segmental duplication, insertions and deletions – there is a good chance that these new data will improve our complex disease mapping efforts. I am quite confident that CNVs are not randomly distributed in the genome
CNV genes encode disproportionately large numbers of secreted, olfactory, and immunity proteins, although they contain fewer than expected genes associated with Mendelian disease […] Natural selection appears to have acted discriminately among human CNV genes. The significant overabundance, within human CNVs, of genes associated with olfaction, immunity, protein secretion, and elevated coding sequence divergence, indicates that a subset may have been retained in the human population due to the adaptive benefit of increased gene dosage.
There is a good chance of retrieving even posthoc CNV information from SNP arrays by taking into account relative signal intensity. Yea, yea.
Addendum
The mouse data are now also online.
CC-BY-NC Science Surf , accessed 06.08.2026