Don´t confuse me with Science of the Surf Continue reading Science of the surf
Do not stand between me and the sun
Diogenes, “the Cynic,” Greek philosopher, was born at Sinope about 412 BC, and died in 323 at Corinth, according to Diogenes Laërtius.
Do not stand between me and the sun – the vitamin/allergy story is getting confusing with the new JACI review. Continue reading Do not stand between me and the sun
Can you see these neat little fingers
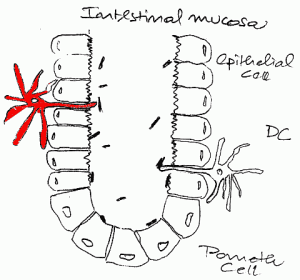
… reaching into the intestinal lumen? They belong to dendritic cells and are depicted also in a nice review about immune responses to commensal and environmental microbes Continue reading Can you see these neat little fingers
A self explanatory letter
Dear Dr. Wjst:
Thank you very much for your letter. However, we don’t allow author commentaries on their own articles, except to supply errata, which is not involved here.
Sincerely, XXX
the dream journal, you name it, yea, yea.
Eat peanut to avoid peanut allergy
There is a new comment in the BMJ about a Lords committee report
a number of recent epidemiological studies had indicated that early peanut consumption in countries such as Israel was associated with a low incidence of peanut allergy in the population. These observations had led many academics to say that exposing a child’s immune system to peanut allergen at an early age might result in tolerance.
It seems that allergen avoidance versus sportively exposure is a never ending story – forth and back and back and forth – and largely irrelevant as being only about the second line of defense?
Of guests and ghosts
Here is a rare occasion where you can identify “guests” and “ghosts” in a paper – a common practice. Continue reading Of guests and ghosts
Look here
From an email that I received today:
while open access has provided the scientific community with broader accessibility, little seems to have been done to make better use of the on-line content. We are trying to address this shortcoming through pubcasts. Pubcasts are 5-10 minute video clips which are integrated with the contents of the open access paper.
I agree with the observation that Open Access hasn´t been very innovative so far in technical terms. On the other hand I feel that my job is less about marketing than development. Are you climbing on this bandwagon? Continue reading Look here
Archie Cochrane speaking
I did not expect what a new Cochrane writes on the prevention of nutritional rickets in term born children — Cochrane Database Syst Rev. 2007 concludes
There a only few studies on the prevention of nutritional rickets in term born children. Until new data become available, it appears sound to offer preventive measures (vitamin D or calcium) to groups of high risk, like infants and toddlers; children living in Africa, Asia or the Middle East or migrated children from these regions into areas where rickets is not frequent. Due to a marked clinical heterogeneity and the scarcity of data, the main and adverse effects of preventive measures against nutritional rickets should be investigated in different countries, different age groups and in children of different ethnic origin.
May I summarize: 1. only a few studies; 2. vitamin D OR calcium and 3. to high risk kids only. This looks different to what Nestlé manufactures at the moment.
Original research in blogs
It does not seem very unusual to have original research in blog posts. Evolgen is doing that by currently publishing a series exploring the evolution of a duplicated gene in the genus Drosophila. So finally science is more than knowing which is a high impact journal – peer review may be replaced comments below.
What’s in your genome?
My latest idea is to create a wiki like annotation server that lets everybody create rules how to analyze an individual genome – we could use the CV dataset as a testbed.
Maybe we should start with SNPs only and develop some ground rules first at which threshold any predictive rule may be applied?
Otherwise these personal genomes will be quite useless, yea, yea.
Auto desensitization
Blackley found already in 1873 an interesting explanation of the “no allergy in farming children” effect by referring to some kind of auto desensitization in this particular environment – e.g. the high pollen and LPS exposure.
Do you know that a commercial allergen preparation used for desensitization already includes a LPS derivate, 3-o-desacyl-4′ monophosphoryl lipid A as an adjuvant? It is believed to push the pollen reaction into a IL12 – IFNg – Th1 pathway. This therapeutic approach already perfectly fits the early explanation of Blackley.
When will the allergy farming lobby ultimately close their files?
Henryk Goldszmit (Janusz Korczak)
moblog – When being in Warsaw for the first time I wanted to honour Henryk Goldszmit / Janusz Korczak by visiting the orphanage. This was a home for Jewish children in the Jaktorowska (former Krochmalna) Street. Henryk, born 1878 or 1879, a physician, writer and outstanding pedagogue was the director of the orphanage since 1913. Continue reading Henryk Goldszmit (Janusz Korczak)
Contrary or Contradictory
A forthcoming “Perspectives in asthma” paper in “JACI” by Litonjua and Weiss will be about the vitamin D hypothesis. Although the authors find that
Evidence exists that vitamin D induces a shift in the balance between TH1 and TH2-rype cytokines toward TH2 dominance
they make a largely unexpected turn by saying that vitamin DEFICIENCY may be to blame for the asthma epidemic. The basic argumentation is
We hypothesize that as populations grow more prosperous and more Westernized, more time is spent indoors and there is less exposure to sunlight leading to vitamin D deficiency…
Given the immediate and effective vitamin D production in skin and its longterm availability by fat stores I cannot follow their last conclusion. Continue reading Contrary or Contradictory
The history of vitamin D discovery, industrial production and marketing
There is a new German dissertation about Vigantol (R) excellently written by Jochen Haas and just published at Wissenschaftliche Verlagsgesellschaft “Vigantol, Adolf Windaus und die Geschichte des Vitamin D”. Altogether 425 pages, it contains a biographical sketch of Windaus (p 32-83), a detailed summary of the juristical questions about irradiating ergosterin (p 96-154), a detailed history of the pharmaceutical production (p 155-238) and finally a chapter about marketing of different brands by Merck (p 238-281). Continue reading The history of vitamin D discovery, industrial production and marketing
I am thrilled to announce
Getting the right expression is not always easy if you are thinking in a different language. “Borrowing good English” as confessed now by a Turkish scientist is therefore understandable. Best regards to Mark Twain from an awful German language speaker.