–Day 2 of Just Science Week–
Most people think that human genes are static entities inherited from generation to generation. They may be right, there are no jumping genes in humans.
In 2000, when defending my thesis in epidemiology, I even had to answer the question of the faculty: “How can allergy have a genetic cause as most allergy cases date back only 1 or 2 generations?”. I explained the concept of susceptibility genes (that were always there) plus some new environmental risk factor (that came in only recently) and passed the colloquium.
Maybe this concept was not completely wrong. By today, however, I could offer more explanations – human genes are on the move and even within 2 or 3 generations. You may still wonder – are we talking about T cell receptor recombination? Yes, this may be a possibility, but not a really new one. More noteworthy are (1) abolished purifying selection (2) population admixture and (3) increased spike in mutations. These are all are independent paths that may combine freely.
Lets start with “abolished purifying selection”. At the beginning of the last century there were much larger family sizes and a much higher infant mortality. In Europe, mortality under the age of 5 has been about 250/1.000 live born children in 1900. It dropped to 50/1.000 around 1950 and is now about 5/1.000 – the effect of improved sanitary conditions, vaccines and antibiotics. Geneticists would describe it as a reduced selection – giving immediate rise to some variants in the gene pool.
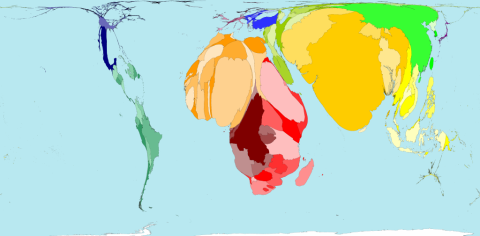
The figure is © Copyright 2006 SASI Group (University of Sheffield) and Mark Newman (University of Michigan). Thanks to John Pritchard from the Worldmapper Team to let me post it here. Territory size shows the proportion of infant deaths during the first year of their life in 2002
Second, think of population admixture. This usually refers to the composition of a population by descendants of a few founders. Except for major migration periods population composition has been kept rather stable over centuries which can be nicely seen in humans living in isolation and having enriched some diseases – examples from Finland, Hutterities, South Tirol, East Adria or Iceand. With current decrease of admixture also the prevalence of diseases frequently seen in these populations will go done. However, some people even expect that other diseases will rise – as unusual allelic variants will meet other unusual allelic variants (which has not been balanced before). This theory is still vague but has interesting aspects that may be followed up.
A third possibility why human gene variants may change within short time comes with the ever increasing age of fathers at reproduction. I came across this only very recently by a paper of Ellegren. With each additional year of the father the number of pre-meiotic cell divisions increases – leading to a permanent (germline) increase of mutations. Most of these will be irrelevant but some may spike in random genes leading eventually to health effects. Crow asks: “Is this a problem? Surely it will be eventually, but probably not immediately”.
Human genes are therefore on the move, yea, yea.
CC-BY-NC Science Surf
accessed 11.03.2026