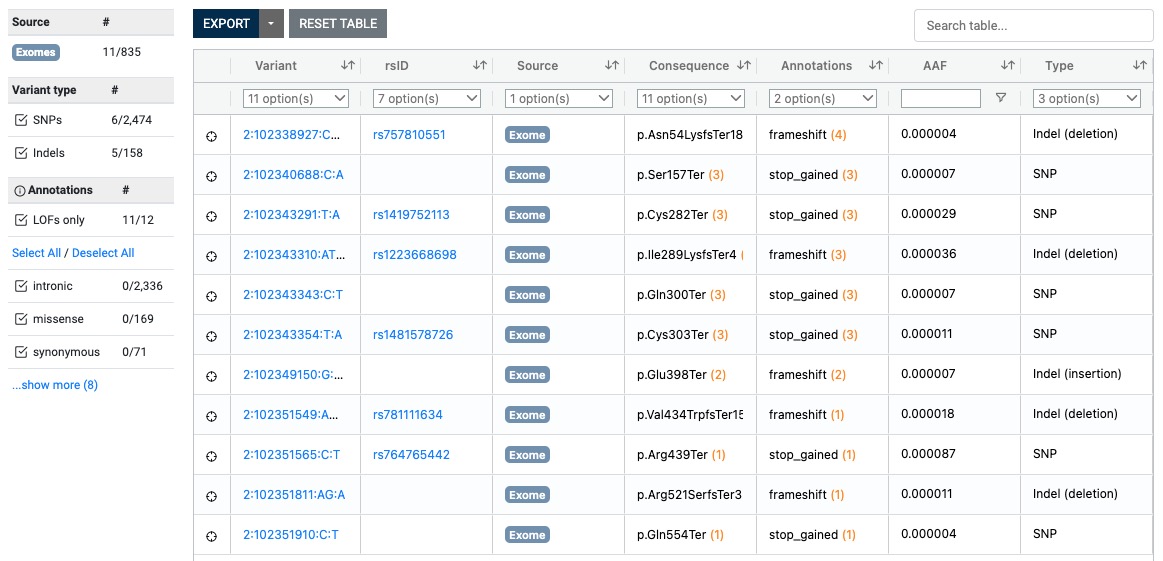
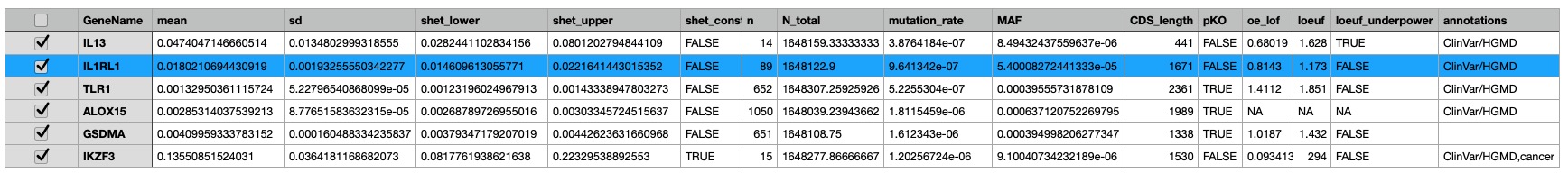
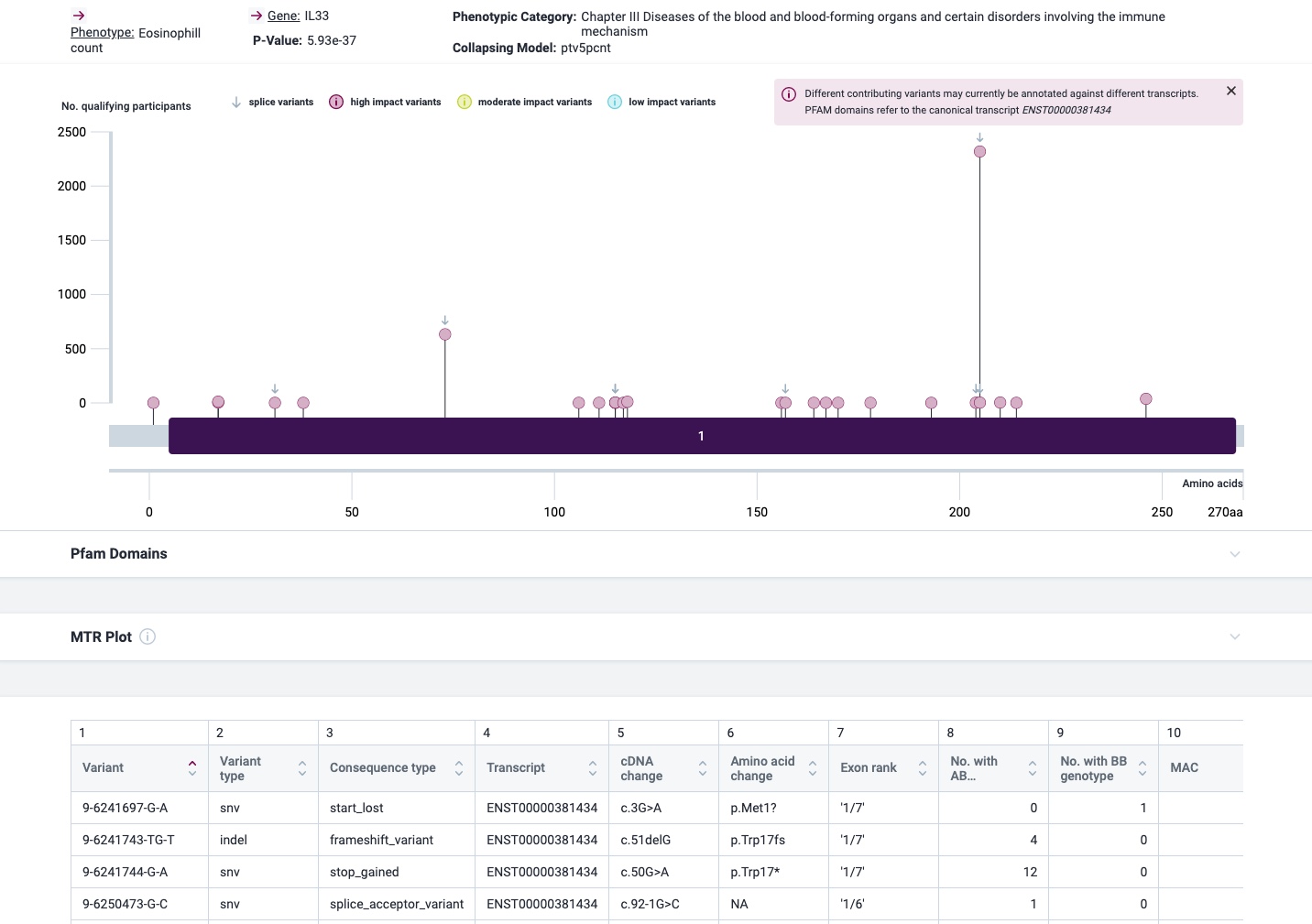
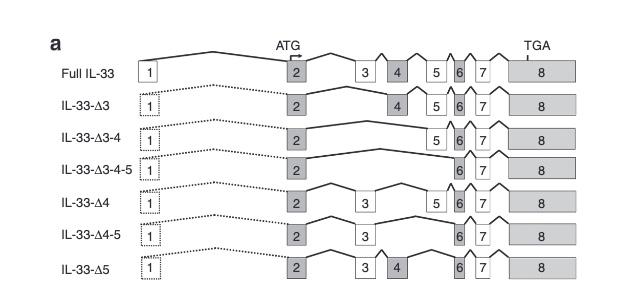
Here is a quick update on some genes of my recent asthma exome paper coming now from the 1 M exome paper published yesterday as a preprint.
loss of function variants IL1RL1. https://rgc-mcps.regeneron.com/gene/IL1RL1, 12 May 2023
Also ClinVar shows that the IL33 receptor is not “essential” making anti IL33 receptor antibodies like etokimab, itepekimab, tozorakimab a safe therapy although not being effective in any LOF mutation carrier.
The most interesting thing in the preprint is in supplemental table 2 with the s-het values for 16,704 genes. From that table I have selected my favorite target IL33 receptor together with TLR1, ALOX15, GSDMA, IL13 and IKZF3 ( BTNL2 could not be found in the list).
asthma exome https://rgc-mcps.regeneron.com/gene/IL1RL1, 12 May 2023
IKZF3 would be dangerous to be touched (see my 2008 commentary) while in the 2022 exome paper I also found only protective variants in the 5′-UTR but not any LOF variant – probably as IKZF3 is the only essential gene in the list.
So what's next? I am still thinking how to reduce my exome set to the causal variants as half of the mutations are probably LD artefacts. And well, it would be super interesting to examine now two extreme inbred populations for their mutation spectrum, loosing either asthma variants by healthy (Amish) or diseased founders (Tristan da Cunha). Unfortunately there is little hope that this will happen – current science is built more on competition than collaboration.
It seems that the respiratory tract isn’t so much influenced by rare gene variants but that there is a strong effect in the immune system.
And there is another interesting fact.
…Surveying the contribution of rare variants to the genetic architecture of human disease through exome sequencing of 177,882 UK Biobank participants …if we look at the …. European population who are carriers of a filaggrin (FLG) PTV, we find those carriers have significantly higher risk for well-known associations, such as dermatitis … and asthma … Concomitant increases in vitamin D levels suggest … increased sensitivity to ultraviolet B radiation.
So far, I have only assumed an asthma/allergy priming effect of oral vitamin D in the newborn gut. This paper now argues for an increased vitamin D sensitivity also in the skin of FLG dermatitis patients which is interesting given the largely contradictory data of serum vitamin D and atopic dermatitis. Maybe dermatologists should focus their research more on skin and local vitamin D turnover?
-II-
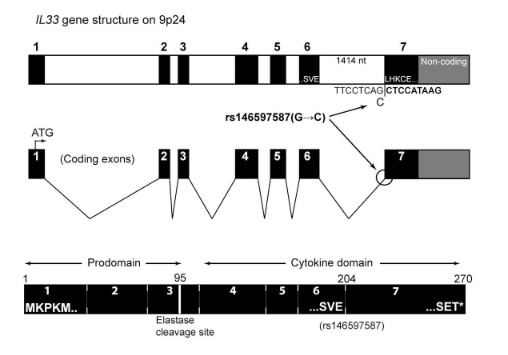
The most prominent IL33 variant carried by over 2,300 people is splice acceptor 9-6250473-G-C followed by 600+ individuals with splice donor 9-6250600-G-T.
There are not too many carriers of this variant by the sheer amount of 177,882 participants. We nevertheless know already something about the seven IL33 splice variants since 2012.
Novel Splice Variants of IL-33: Differential Expression in Normal and Transformed Cells Journal of Investigative Dermatology (2012) 132, 2661-2664; doi:10.1038/jid.2012.180
Fig 3A Smith et al. A rare IL33 loss-of-function mutation reduces blood eosinophil counts and protects from asthma, PLoS Genetics 2017 – describes the splice site as NM_001199640:exon7:c.487-1G>C or rs146597587-C
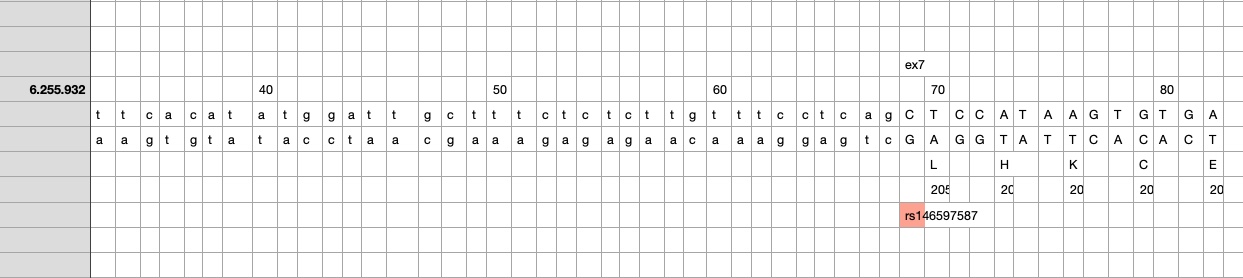
So I did a sequence match to compare the new finding with these older publications.
own sequence match exon7 using data from dbSNP, UCSC GoldenPath and Uniprot – reference is hg19
Indeed, the 2017 paper already described rs146597587 which is probably identical to the splice acceptor 9-6250473-G-C in Astra UK Phewas (genome positions do not match – I used hg19 while I don’t know the Astra reference) . Astra says also c.613-1G>C while rs146597587 is just upfront of my codon 205 (3*205=615) whatever that means.
The Astra UK Phewas at least confirms the Iceland paper above
rs146597587-C associates with lower eosinophil counts (ß= -0.21 SD, P = 2.5×10-16, N = 103,104), and reduced risk of asthma in Europeans (OR = 0.47; 95%CI: 0.32, 0.70, P = 1.8×10-4, N cases = 6,465, N controls = 302,977). Heterozygotes have about 40% lower total IL33 mRNA expression than non-carriers and allele-specific analysis based on RNA sequencing and phased genotypes shows that only 20% of the total expression is from the mutated chromosome. In half of those transcripts the mutation causes retention of the last intron, predicted to result in a premature stop codon that leads to truncation of 66 amino acids.
So it is basically a rediscovery meaning that we reached saturation.
This something that I always avoided in human research – blaming genes for resistance to environmental stressors.
Nevertheless a Californian group (https://doi.org/10.1371/journal.pgen.1008528) now tested 101 mouse strains for lung resistance with exposure to diesel exhaust particles (DEP). After sensitizing the animals with dust mite and aluminium they could also test metacholine hyperreactivity (AHR).
Strains that exhibited the highest lung resistance after control exposure were not necessarily the same as those with high lung resistance after DEP exposure. It is unclear which strain was used for the consecutive GWAS. Did they put all mice into one cage for that?
The metacholine AHR GWAS results are not very impressive. And there seem to be also errors, as for example the lead SNP on chr 19 (rs51547574, near IL33) is shown with different allele frequencies in text and Fig 2. As the expression quantitative trait locus (eQTL) for Il33 is not in the lung, I think there is nothing to memorize here – IL33 is just a gatekeeper for surface integrity.
In a next step I wouldhave expected a GWAS for resistance change after DEP but FIG 3 only gives the result of Δ AHRDEP-AHRPBS data at an abitrary methacholine dose of 10mg/ml. The identifed locus could be interesting but as the LD there is rather high without any corresponding eQTL (I always wondered why there has never been a significance threshold for LD blocks, only for isolated SNPs?), the logic of the paper is broken here. Induction of lung resistance by DEP was significantly blunted in Dapp1-/- female mice? What about male Gm5105-/-, Mttp-/-, and Lamtor3-/- animals?
Hopefully nobody else will now try to find diesel, ozone, NOx resistance genes in humans as this is not a a scientific but a political issue…
These reviews do not tell you so much about the regulation while regulation has recently elucidated by Gour et al. who describe a tropomyosin-dectin-1 interaction of the human host. Why is tropomyosin such a frequent target of human IgE?
Muscle protein tropomyosin is an important IgE target in a number of nematode infections; Onchocerca volvulus ; Ascaris lumbricoides; Anisakis simplex; and tropomyosin from the blood fluke Schistosoma mansoni is also a human IgE antigen. Tropomyosin is highly conserved across many invertebrates and is responsible for much of the IgE cross-reactivity between Ascaris and dust-mites.
I haven’t found any good answer to this question. As tropomyosin affects contractility – this seems like “shooting into the leg” of worms whenever they attempt to invade.
Maybe Gour et al. did not know the earlier dissertation from Berlin that already showed a reduced inflammation in the OVA mouse model by administration of recombinant tropomyosin.
The broad cross reactivity to tropomyosin gives rise to the question if helminth tropomyosin could induce allergic reactions to itself and/or tropomyosin of different organisms. Considering the fact that filarial nematodes express tropomyosin on their surface […] and that the continuing turnover of microfilariae confronts the host with relevant amounts of tropomyosin makes this question even more appropriate.
Worms seems to be attacked by anti-worm-surface-tropomyosin IgE whenever the worm tries to invade the epithelium during an acute infection. During invasion extracellular IL33 is cleaved into a shorter form with enhanced activity attracting more immune cells.
During chronic infestation nothing happens as long as the worm does not invade and doesn’t trigger any IL33 alarmin. As there is continuous tropomyosin antigen antigen contact, the host is slowly desensitzed, clearing IgE in favor of IgG4.
Is this also a model that explains allergy? We don’t know the details but maybe this antigen recognition / response system is being disturbed where allergens like Der p1 mimicking a worm infection by tropomyosin can trigger the allergic reaction in particular as Der p1 a cysteine protease also mimicks an invasion signal.