Following several unsuccessful attempts to implement a parallel computing platform for R statistical software, I am showing here my current approach that is largely influenced by a recent paper on cluster programming in c’t 6/06 by Oliver Lau (sorry, no online version). My primary interest is with the R library snow (or snow-ft) that offers the function clusterApplyLB. This function is all I need for my R programs.
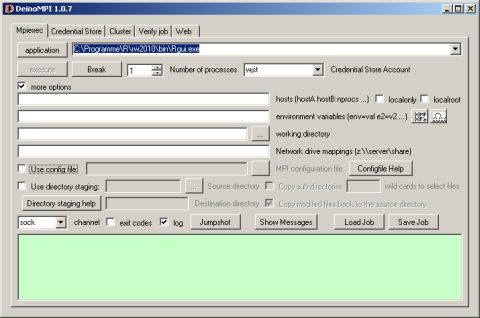
Now it gets more complicated: library(snow) depends on library(Rmpi): Hao Yu has an excellent description at www.stats.uwo.ca/faculty/yu/Rmpi how to set up the mpi layer with MPICH2. I am currently experimenting with DeinoMPI a closely related high performance Windows interface. According to its developer David Ashton it has the following advantages
First, DeinoMPI does not require MPI applications to be started by mpiexec in order to call MPI_Comm_spawn so you could load Rmpi from the Rgui.exe without having to bother with calling mpiexec. Second, DeinoMPI loads the user profile when starting applications so if you query the user’s temporary directory you will get the user specific path and not the Windows system temp directory. Third, DeinoMPI handles arguments with spaces correctly if you quote them so you can pass environment variables with spaces in them. Fourth, DeinoMPI allows you to use the MPI Info object to pass extra options to MPI_Comm_spawn like drive mappings. So you could create an MPI_Info object and set wdir=z:\ and map=z:\\server\share. Then pass this info object in with the MPI_Comm_spawn command and you could map a network drive and launch an executable from this drive.
So far the Rmpi package is compiled for MPICH2 (not DeinoMPI) so it won’t run with only DeinoMPI installed but there is a good chance that this will change in the near future.
Further useful references are in the R newsletter 2003, p21 cran.r-project.org/doc/Rnewsand a paper in the UW Biostatistics Working Paper Series on “Simple Parallel Statistical Computing in R” by Anthony Rossini and LukeTierney.
BTW, haplotypes of the hapmap project were computed on a 110 node cluster provided by both Peter Donnelly’s Mathematical Genetics Group www.stats.ox.ac.uk based at the Oxford Centre for Gene Function and by a 128 node compute cluster provided by the Oxford e-Science Centre e-science.ox.ac.uk as part of the National Grid Service[to be cont’d…].