The November AJHG has an excellent re-analysis of the dysbindin-schizophrenia association using new methodology that surpasses all previous meta-analysis techniques. As the single SNP association results from the previous 6 studies cannot be directly compared, they construct a European super-hap map from all tag SNPs in that region, place them in a phylogenetic tree before finally mapping all single associations on these haplotypes. Their Fig.1B show the main results; as the circles in Fig.1B are somewhat confusing, I have withdrawn their results – adding the haplotype frequencies and ordering the studies by year of publication.
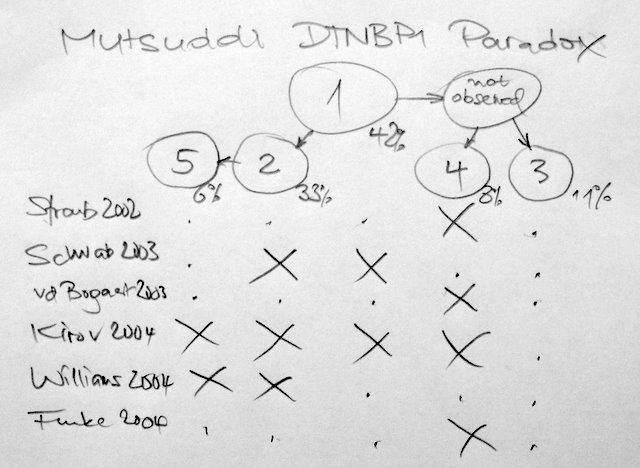
We may think of a triple-blind study – neither patients, nor PIs, nor we did know anything before. The results are alarming. I do not understand how the Kirov set could have included all haplotypes and why the Schwab/Williams set is in opposition to the Straub/Bogaert/Funke set.
What could have gone wrong? The authors of the current re-analysis believe that population differences are an unlikely reason for the inconsistency as the allele frequencies match between studies. Good news that genotyping errors may be largely excluded.
Unfortunately the authors remain vague why there is no common causal variant. Have there been different sampling schemes, different diagnostic thresholds, different environmental exposures in the previous studies? Is dysbindin at all a schizophrenia gene, or only under a certain genetic background? It seems possible that studies of one branch are false positives. Or is the haplotype reconstruction in the re-analysis erroneous for whatever reasons?
Von Münchhausen is well know for escaping from a swamp by pulling himself up by his own hair. I would like I could do that too.