There were some diabetes experts at my former work place who opposed against doing COVID19 studies even sending us epidemiologists into home office while leaving a large study in Munich to the police, BMW and medical students…
Funnily enough, they even warned about any COVID19 research in Cell magazine
… widespread redeployment of world-class expertise from other areas into the current acute phase of COVID-19 research can lead to substantial loss of focus. A longer-term diversion of resources runs the risk of stifling much-needed new basic research and technological breakthroughs that have the potential to revolutionize biomedicine. … A refocus on COVID-19 research activities is likely to engender adverse effects on society’s clear and urgent need for sustained research into major diseases, which will continue to afflict humankind and remain leading causes of death and disability well beyond the end of the acute COVID-19 challenge.
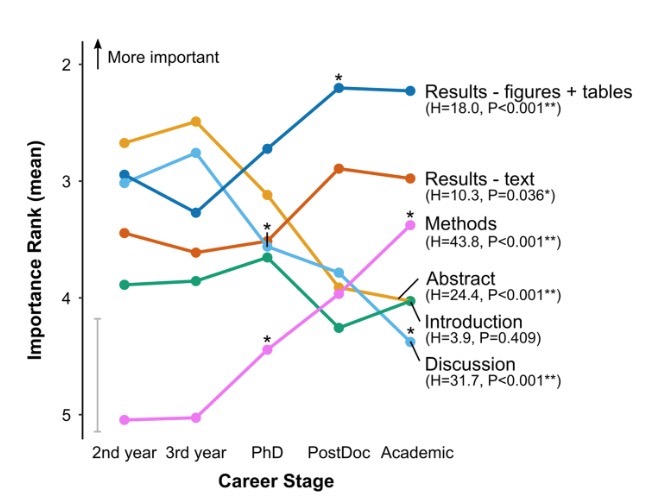
So we had plenty of time to see studies coming up that loss of focus COVID19 is now even inducing their major disease – early onset of diabetes. The story now goes on to a JAMA metaanalysis and a Nature commentary that confirms the diabetes association in 42 studies including 102 984 youths and showing a
higher incidence rate during the first year of the pandemic compared with the prepandemic period (incidence rate ratio [IRR], 1.14; 95% CI, 1.08-1.21). There was an increased incidence of diabetes during months 13 to 24 of the pandemic compared with the prepandemic period (IRR, 1.27; 95% CI, 1.18-1.37).
The accompanying commentary makes a direct damage of SARS-CoV-2 to pancreatic cells in children unlikely while the most
probable explanation is that the immune system’s attack on the pancreas is triggered by a COVID-19 infection, which happens with other infections as well, like enteroviruses and hepatitis B.
I can agree on that while my question now: Do genes play a major role?
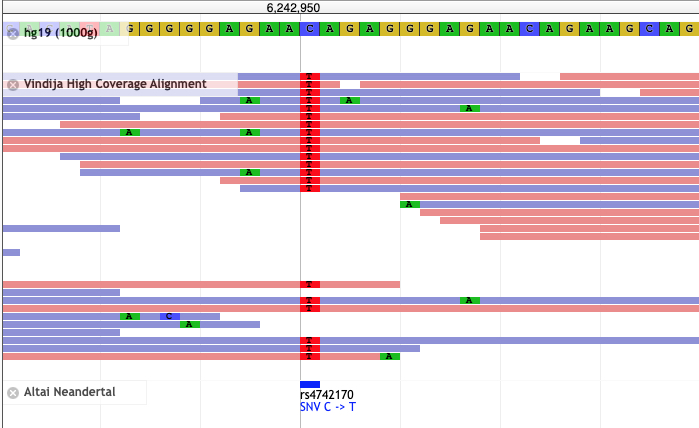
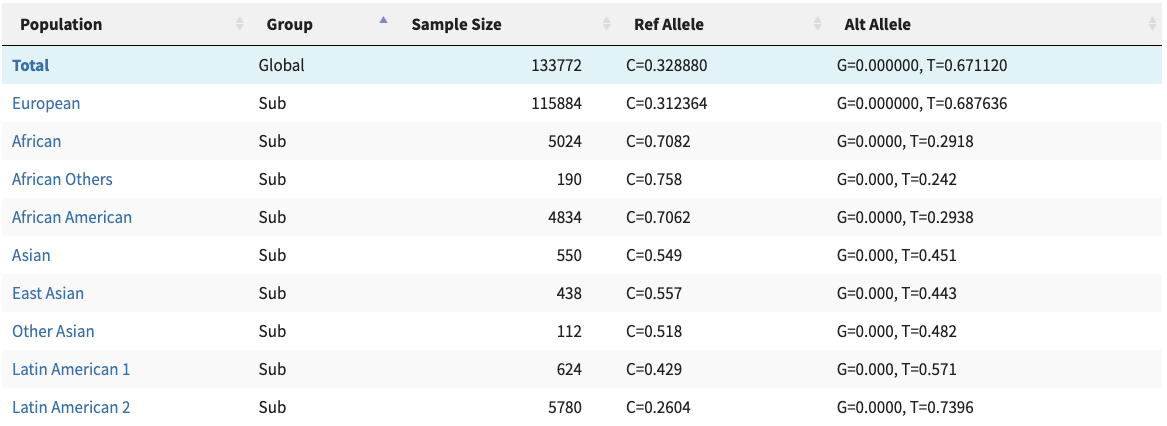
Most genetic research of COVID-19 went into disease severity while there was a paper last week describing disease protection by HLA-B*15:01. As summarized in Science this study of blood donors infected by seasonal coronaviruses showed that
T cells from the subjects with HLA-B*15:01 also reacted aggressively to a fragment of the spike protein from two of the seasonal coronaviruses. This fragment is almost identical to the snippet of SARS-CoV-2’s spike protein that the researchers had tested. The results suggest that when people with the HLA-B variant came down with colds caused by seasonal coronaviruses, they obtained a degree of immunity against similar coronaviruses, including SARS-CoV-2.
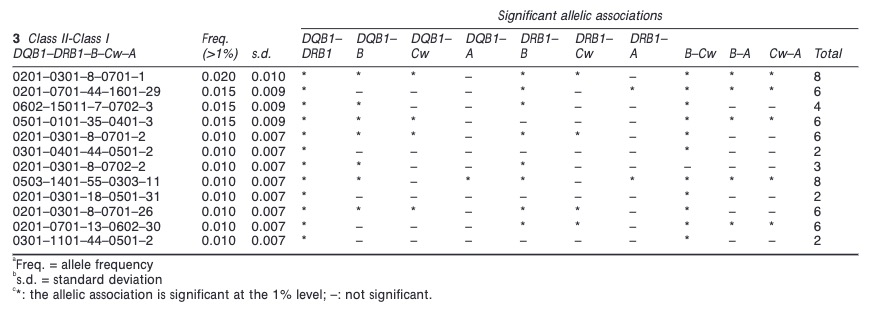
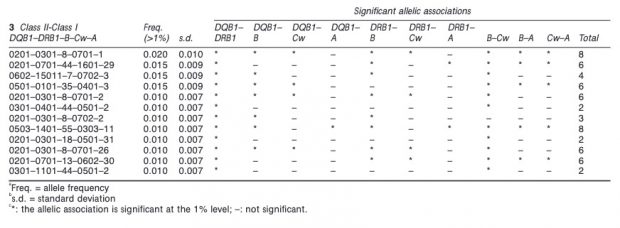
For type 1 diabetes an association with HLA DR/DQ has been described not only be us but also by many other groups before.
This association is however with class II, not class I as found now in COVID-19. Nevertheless there could be some linkage disequilibrium between class I and II variants although I could not find the B*15 allele in an earlier paper.
Or is the T1D/COVID119 association just by co- or cross activation of the immune system?
Before any expensive profiling of antibodies it may be worthwhile to go for some association testing eg HLA-B*15:01 in T1D as there are known complex transcriptional regulatory circuits in the HLA locus.
Research strategies should never be dogmatic…
 “Genetic drivers of heterogeneity” is the new description for the failed genetic concept of “reverse genetic engineering” – where we see now only sand running through the fingers.
“Genetic drivers of heterogeneity” is the new description for the failed genetic concept of “reverse genetic engineering” – where we see now only sand running through the fingers.